
经导管主动脉瓣产品国际监管案例及其启示
2021-12-02 07:20王泽华盛恒松滕颖影
精准医学杂志 2021年5期
王泽华 盛恒松 滕颖影
(国家药品监督管理局医疗器械技术审评中心,北京 100081)
目前,全球范围内罹患心脏瓣膜疾病患者的数量较多[1],在中国约有2 500万例,位居心血管系统疾病的第3位,其中以风湿性心脏瓣膜病居多,其次是退行性心脏瓣膜病,近几年也呈逐年递增趋势[2]。通过介入方法治疗各类瓣膜病变已成为心血管疾病治疗领域发展最为迅速的方向[3]。随着介入医疗技术、创新器械开发和材料处理工艺的不断进步,心脏瓣膜疾病介入治疗的适用证不断拓宽,已逐步从最初的外科禁忌证患者到外科高危患者,又进一步拓展至中低危患者[4-7]。适应证的改变,意味着介入治疗瓣膜疾病的市场在进一步扩大,随着商业化瓣膜产品的广泛应用,瓣膜植入物失效的风险也逐渐显现出来。本文在充分参考了美国已上市经导管主动脉瓣膜的注册情况和上市后的风险报道及管理经验基础上,对国内同类器械的监管进行思考,并提出初步建议。
1 心脏瓣膜疾病的介入治疗概况
人工心脏瓣膜的研发始于20世纪40年代后期,随后几十年间,机械瓣和生物瓣逐渐孕育并研发出来,其在心脏瓣膜疾病治疗过程中取得了不错的治疗效果。经导管主动脉瓣置换(TAVR)手术是主动脉瓣疾病治疗领域的里程碑,与外科主动脉瓣置换术相比,其优势在于减少了手术创伤和创口并发症、最大程度避免了主动脉阻断、更适用于体质衰弱和手术风险高的心脏瓣膜病患者。
自从2002年CRIBIER等[8]首次在人体上完成TAVR手术以来,TAVR手术技术发展迅速,目前在手术风险较高、体质虚弱、瓷化主动脉以及主动脉瓣生物瓣衰败的患者中已经得到广泛的应用。中国TAVR手术起步稍晚,2010年复旦大学附属中山医院在我国进行了首例TAVR手术[9],随后TAVR手术在我国逐步推广应用,我国自主研发的TAVR产品临床应用效果良好[10-12]。结合国外的临床指南,经过多年的临床实践积累,我国也陆续发布了多份适合中国人群的专家共识。专家共识的推广极大地推动了我国TAVR手术的开展和普及,并在一定程度上促进了经导管治疗方式在各个瓣膜部位的应用,为心脏瓣膜疾病的治疗积累了宝贵的经验。
2 美国高风险医疗器械注册监管制度
对于高风险医疗器械的监督管理,美国食品药品监督管理局(FDA)是由医疗器械和放射健康中心(CDRH)主要负责。虽然监管途径众多[13],但主要分两阶段实施监管:①上市前,通过审批计划,加强高风险(Ⅲ类)医疗器械的上市前批准(PMA)审查工作[14],确保创新的、高风险医疗器械的安全有效性。②上市后,通过监测计划、科学研究、法律约束和教育计划等等措施,最大限度地确保上市后医疗器械的安全有效,同时将患者偏好信息也纳入到医疗器械监管流程当中,更多地考虑了患者的使用体验[15]。
正是这一系列监管措施的实施,使得医疗器械在设计、上市使用以及更新迭代的整个的过程中,形成了一整套保证公众用械安全的、完善的统一体系[16]。
2.1
美国《食品、药品和化妆品法》(FDCA)对医疗器械进行了特定分类。对于Ⅱ类和Ⅲ类医疗器械,产品需要分别通过上市前通知(PMN)和上市前批准(PMA)后方可进入市场。对于Ⅲ类高风险产品,在上市前的注册环节,FDA提供了多种途径的审批。FDA的PMA审批制度包括8种类型[17]:除原始PMA外,其余的7类均属于变更类型。根据不同的变更的风险特点确定不同注册路径下需提交的变更证据集,例如涉及全新产品或者已批准产品的重大变更则需重新注册,而仅仅是临床适应证、设计及组成等方面的一般变更、器械的微小设计变更,以及生产过程、工艺、包装及说明书等非重大变更,则可通过变更注册路径提交对应的变更支持证据,以支持注册。
2.2
对于产品上市后的相关风险,CDRH按照流程进行分步实施,逐级排查,并得出最终判断的决策意见。产品上市后的安全监测流程见图1[16]。概括来说,FDA通过对基本信息(不良事件报告、企业核查、召回、上市后研究等过程中产生的数据和信息)的监测,对获批医疗器械在上市后进行安全问题的识别[18]。以监测过程中所识别的安全性问题为基础进行评估,并进一步确认所收集到的医疗器械使用的风险,最终根据具体的风险进行进一步沟通交流、培训或者采取强制性措施,并制定了器械上市后安全管理的标准[18]。该系列过程的实施旨在通过有效地管理和收集数据,得以全过程监测上市后医疗器械的安全性以及有效性,从而最终保证公众的用械安全。

图1 CDRH上市后安全监测流程
3 美国瓣膜产品注册现状
3.1
根据FDA相关法规,瓣膜产品注册路径主要包括PMA和人道主义器械豁免(HDE)两种。其中属于PMA路径下的器械及其分类编码分别如下:瓣膜置换(DYE)、非同种异体心脏瓣膜(LWR)、经导管二尖瓣修复器械(NKM)、经导管主动脉瓣置换(NPT)、经导管二尖瓣假体(NPU)、机械瓣(LWQ)、肺动脉瓣(NJK)以及经导管肺动脉瓣(NPV);属于HDE路径下的器械及分类编码分别如下:同种异体心脏瓣膜(MIE)、经导管肺动脉瓣置换(PAL)和儿科主动脉瓣(PAP)[19]。
3.2
在所获得批准的PMA当中,DYE、NPT、LWR和LWQ类分别占据前四位,且获批数量远高于其他同类产品;同时,变更注册的批准数远高于首次注册的批准数量(图2)。因此足见变更注册途径在整个FDA的PMA注册系统中的重要地位。
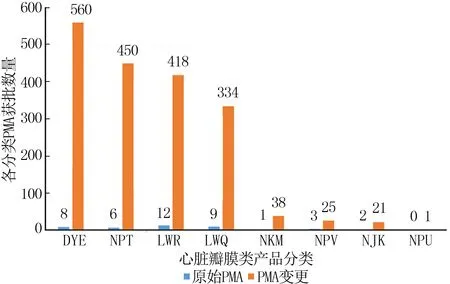
图2 瓣膜产品PMA首次审批与变更审批对比情况
根据数据统计(图3),近30年来,前4类瓣膜类产品的申请量总体呈逐年递增趋势。2013年以前,主要是以机械瓣膜产品DYE为主;2013年以后,经导管介入瓣膜类产品NPT和非同种异体瓣膜LWR的申请获批数逐年提高,并始终维持较高的年批准量。
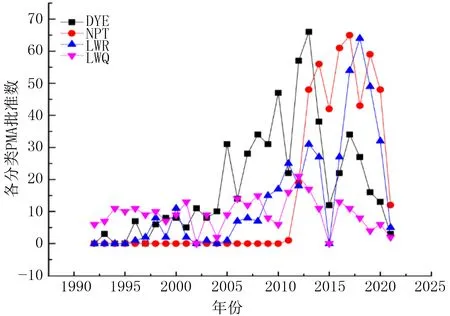
图3 前四类瓣膜产品历年PMA审批次数走势图
NPT是当下医学研究和注册热度较高的产品(图4),首次获得批准的PMA占比仅为1%,其余占比99%的类型为变更批准PMA。在所有的变更批准类型中,最主要的为涉及制造商/灭菌商/包装商/供应商生产过程的工艺类型的变更,所占比例为75%,见表1。在所有变更PMA中,30 d追踪类型的PMA类型占据绝大多数,表明大多数均为轻微变更;此外,工艺变更占据了绝大多数类型。

图4 所批准的PMA类型分布

表1 PMA变更的理由及分布
4 美国瓣膜产品监管现状
瓣膜类产品特殊的应用部位和功能,决定了该类医疗器械一旦出现失效,对患者所造成伤害是比较严重的。FDA特别强调对该类产品的全生命周期的管理。随着该类产品在市场上逐渐推广使用,因进行经导管瓣膜手术而产生的器械安全事件报告逐渐增多。目前FDA主要通过制造商与用户机构设备使用数据库(MAUDE)和全生命周期管理数据库(TPLC)对医疗器械的安全性进行监管。
4.1
根据目前已获批的NPT产品上市后MAUDE汇总[20],见图5,在总体趋势方面各公司产品的不良事件分布比较相似,占据绝大多数比例的为受伤类型,其次为死亡类型,功能失效以及其他类型相对来说比较少。在MAUDE报告当中指出,该类产品不良事件对于患者所造成的相关影响主要为:心律失常、卒中、失血、非特异性心电图改变、完全性心脏传导阻滞、主动脉瓣反流、死亡、心肌梗死以及心房颤动等。

图5 获批PMA的NPT产品上市后累计不良事件数量
4.2
根据CDRH历年统计,相比其他类别的瓣膜类产品,NPT类产品不良事件呈明显上升趋势[21],见图6。NPT产品作为创新产品,自首次获批以来,始终保持了较高的年申请量,其不良事件的发生率随着各产品的上市推广呈现了逐年递增趋势[16],并且尚未有减缓趋势,此类产品的使用风险应该予以重视(图7)。
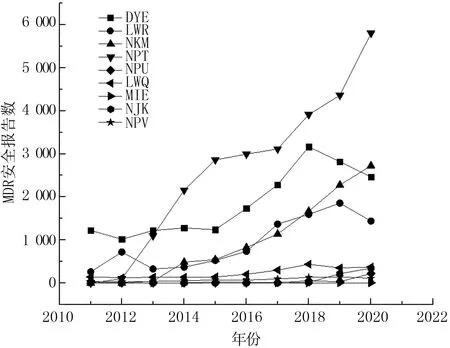
图6 TPLC瓣膜类产品的安全事件报告数量

图7 TPLC的NPT产品PMA与安全事件报告汇总
TPLC报告中相关安全报告显示,对患者的常见伤害有心律失常、外伤伤害、完全性心脏传导阻滞、主动脉瓣反流、死亡、卒中、非特异性心电图改变、血管剥离、血管系统受损、出血、低血压、脑梗死、钙沉积和或钙化等。
根据PMA所批准NPT产品的历年召回统计(图8),相对于其他三类高风险医疗产品,目前该类产品的召回发生率处于相对较低水平,但近几年的召回率呈明显增高的趋势[22],未来应持续关注。召回的原因主要为瓣膜产品的设计、瓣膜输送器械的设计、用于瓣膜压握装载的器械、产品的无菌包装及产品标签等问题。
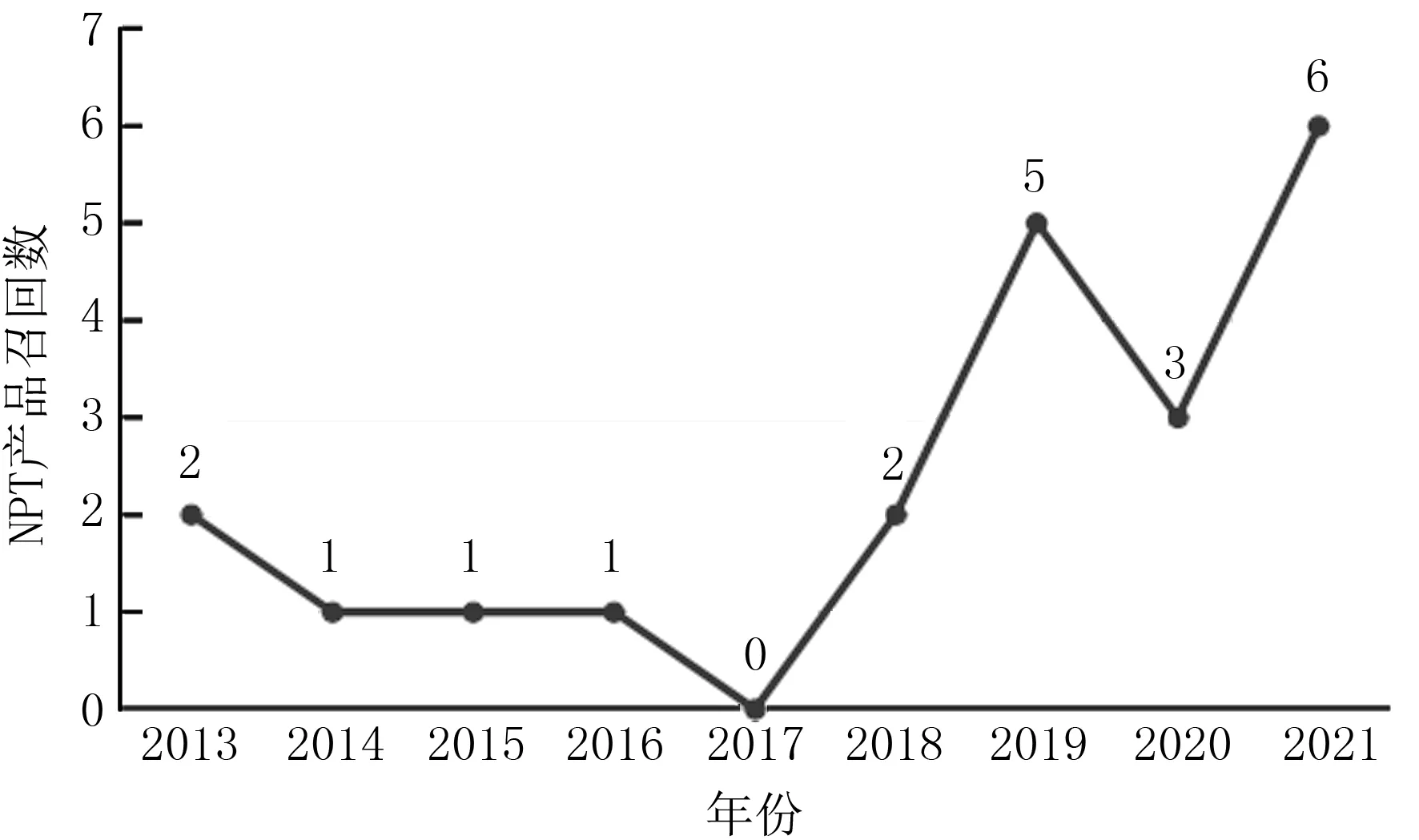
图8 NPT产品历年召回统计
5 瓣膜类产品的监管思考
5.1
由于医疗器械的复杂性和多样化,造成了当前监管工作困难重重,例如制造商和使用机构等上报的MAUDE数据仍可能存在提交数据的不完整、不准确、不及时、重复、未经核实或有偏倚等缺点。对市场监管者而言,如何对所获取数据的真实性、全面性和准确性进行科学判断仍是一项巨大的挑战。近年来,FDA亦在逐步通过一系列改革计划来不断完善其各项监管措施。医疗器械的监管科学是服务于医疗器械监管活动的科学研究[23],循证科学源于循证医学的科学内涵,监管科学和循证科学的有机结合可以促进医疗器械监管水平的进一步提升。
此外,部分行业协会在推动技术进步和监管进步方面也发挥了积极的作用。如美国的STS/ACC TVT Registry注册登记研究是由美国胸外科医师学会、美国心脏病学会联合FDA及医疗保险和医疗补助服务中心(CMS)合作创建的[24-26]。该研究旨在通过标准化的数据收集分析工具,用于监测患者经导管瓣膜置换和修复手术后相关的有效性和安全性。目前,该注册研究已发展成全球最成熟的瓣膜注册研究之一,也成为了国家监测计划的重要组成部分。经过数十年发展,美国全境已有超过780家医院加入了该研究计划,该计划已在医院手术质量反馈和使用指南的更新、国家承保的医保福利的保障、FDA的上市后报告、临床证据的产生等多方面发挥着积极的作用。
5.2
中国瓣膜领域的创新活动异常活跃,无论是进入创新通道的器械数还是获批准上市的创新器械数均列所有创新器械分类的前茅[27]。随着产品不断地上市应用,国内瓣膜领域的监管经验也在不断积累完善,每年国家和省级医疗器械质量抽查检验也在一定程度起到了督查和警戒作用。目前,国内已上市多款瓣膜类产品,注册证备注栏中要求对其远期安全性和有效性的进一步确认。针对目前已获批的经导管主动脉瓣系统,建议在上市后完成对上市前临床患者的继续随访研究及真实世界的中远期研究等[28]。
此外,由于中国人群主动脉瓣钙化更为严重,存在二叶瓣疾病比例较高等的现状[29],且国外当前的临床经验与国内的临床经验有差异,因此建立符合我国诊疗实际的临床注册登记数据库具有重要的临床意义。通过建立相关数据库推进我国人群主动脉瓣临床大数据的研究,可以客观分析和比较批准产品的临床应用情况,有利于上市器械设计得到持续改进,也有利于积累关于患者器械型号的选择和不同人群受益风险分析等方面的信息,为优化手术操作、患者术后服药及康复指导分析等过程提供更多数据支持,从而更好地优化该类产品的长期临床表现(如降低血栓发生率、提高瓣膜耐久性等),推动高发不良事件等问题的解决。
总之,采用临床注册登记数据库的方式进行临床数据的管理是一种值得借鉴的方式,其目的在于切实贯彻落实产品全生命周期的风险管理理念,吸收借鉴他国经验,优化应用,建立适合我国国情的更加科学规范的监管方式,从而最终能更好地促进整个医疗器械产业的良性发展,更好地保证全社会及时获得优质安全的医疗器械服务,更好地保证人民群众的生命安全。
猜你喜欢
护理与康复(2022年6期)2022-11-25
康复(2022年25期)2022-10-05
右江医学(2022年7期)2022-08-07
中国典型病例大全(2022年9期)2022-04-19
影像研究与医学应用(2021年17期)2021-10-18
临床超声医学杂志(2021年2期)2021-03-02
大学生(2017年10期)2017-10-23
中西医结合心血管病电子杂志(2015年10期)2015-10-21
意林(2015年20期)2015-10-21
成都体育学院学报(2014年1期)2014-07-24
