
基于微液节点采样的定量质谱成像技术在包装材料中光引发剂定位与检测中的应用
2021-11-28 05:26梁秋菊黄宇轩李优梅王志国
分析测试学报 2021年11期
梁秋菊,吴 倩,黄宇轩,李优梅,王志国,杜 文
(1.湖南中烟工业有限责任公司技术中心,湖南 长沙 410007;2.中南大学 化学化工学院,湖南 长沙 410083)
目前,标准的光引发剂检测方法主要有气相色谱-质谱法和液相色谱-质谱法等[4-7]。但这些方法在检测前需进行纸张破碎、大剂量的溶剂提取以及液液萃取和固相萃取等繁琐的样品前处理过程,然后再进行数十分钟到1 h的色谱-质谱检测。这些繁琐的样品处理过程是质谱分析速度慢、通量小的主要原因。同时,这些过程还会引入更多的检测误差和样品分解,使检测结果不准确。另外,出于印刷效果的要求,包装材料的某些重点部位可能会使用丝印或胶印技术,而传统检测方法的大面积采样方式对这些重点部位有稀释作用,因此有可能出现采用标准方法无法检出局部超标或超范围使用光引发剂的可能。
为了提高质谱分析的速度和检测的准确度,原位质谱分析技术近年来发展快速。常见的原位质谱分析有解析电喷雾质谱(Desorption electrospray ionization,DESI)[8]、实时直接分析质谱(Direct analysis in real time,DART)[9]、激光烧蚀电喷雾电离(Laser ablation electrospray ionization,LAESI)[10]、微液节点采样技术(Liquid microjunction surface sampling,LMJSS)[11]等。这些技术不但可以原位快速获得样品中分析物的相对含量,同时由于采样探针的空间分辨率,也可得到分析物在样品表面的空间分布信息,为分析检测提供更丰富的信息。这些技术各有优势,其中微液节点采样技术装置简单、成本低,且可原位得到液态样品,使得其可实现采样后加标或与其他分离技术联用,方法灵活多变[12]。但对于固体样品的原位分析,目前这些方法存在定量困难以及定量结果不具代表性等问题。这是因为这些原位电离方法通常只能对微小的区域进行非消耗性的采样,使得采样效率很低且难以预知。同时,由于固体样品难以进行均匀的加标,因而无法通过标样实验得到绝对的采样效率并进行定量校正。因此,甚少有研究采用这些原位质谱方法进行绝对定量分析。
近期,本课题组针对光引发剂的原位定量分析问题开发了微液节点采样-质谱检测方法,并通过动力学校正法有效地解决了光引发剂原位定量的问题[13]。但该方法主要适用于一定面积内光引发剂的原位定量,要将方法应用于光引发剂成像分析还存在空间分辨率低、检测时间长和操作复杂的问题。本研究在前期工作的基础上开发了适用于光引发剂定量质谱成像的微液节点采样-质谱分析方法,将其应用于包装纸中多种光引发剂的定量成像中,并采用传统气相色谱-质谱法分析验证方法的准确性。本方法可为包装纸中光引发剂的检测提供更准确的空间分布信息以及绝对定量信息,为包装纸中光引发剂的质量监控提供技术支撑。
1 实验部分
1.1 仪器与试剂
电喷雾离子源-离子阱串联飞行时间质谱(IT-TOF,日本岛津公司);SCION气相色谱-质谱联用仪(456-GC-SQ,天美有限公司)。
乙腈、甲酸均为色谱纯,购于默克公司(Darmstadt,德国);纯净水购自杭州娃哈哈公司。4-(二甲氨基)苯甲酸乙酯(EDB,99.5%,10287-53-3)、对二甲氨基苯甲酸异辛酯(EHDBA,97.0%,21245-02-3)、苯甲酰甲酸甲酯(MBF,97.0%,15206-55-0)、安息香二乙醚(BDK,100%,24650-42-8)、邻苯甲酰苯甲酸甲酯(OMBB,100%,606-28-0)、2-羟基-2-甲基-1-苯基丙酮(PI1173, 99.4%, 7473-98-5)、二 苯 甲酮(BP, 100%, 119-61-9)、2/3/4-甲 基二 苯 甲酮(2/3/4-MBP,97%,131-58-8/634-65-2/134-84-9)、1-羟基环己基苯基甲酮(PI 184,99.4%,947-19-3)、2-甲基-1-(4-甲硫基)苯基2-吗啉基-1-丙酮(PI 907,98.9%,71868-10-5)、2/4-异丙基硫杂蒽酮(2/4-ITX,100%,5495-84-1/83846-86-0)、4-苯基二苯甲酮(PBZ,100%,2128-93-0)、2,4-二乙基硫杂蒽酮(DETX,99.2%,82799-44-8)、4,4'-双(二甲氨基)二苯甲酮(MK,100%,90-94-8)和4,4'-双(二乙氨基)二苯酮(DEAB,100%,90-93-7)18种光引发剂单标标样购于AccuStandard(NewHaven,美国),溶于乙腈配成1 mg/mL混标,于-20℃冰箱中储存备用。
包装纸购于超市。
1.2 实验方法
传统定量方法:准确裁取主包装面,面积取10.0 cm×5.0 cm;将裁取的0.5 dm2试样剪成约0.5 cm×0.5 cm的碎片,进行后续分析。
台湾应用型本科大学对学生的评价机制灵活多样,评价主体多元化,评价方法多样化。不同的课程有相应的评价标准,以实践课程为例,学生实习单位工作评分占60%,老师教导占10%,实习作业与心得报告占30%。
质谱成像方法:直接取包装纸的主包装面,将其固定在样品台上直接进行成像分析。
1.2.1 包装纸中光引发剂的传统定量检测根据烟草企业标准方法YQ/T 31-2013《卷烟条与盒包装纸中光引发剂的测定气相色谱-质谱联用法》[14]操作:将剪碎的试样置于50 mL具塞三角瓶中,加入20 mL水后静置30 min,再准确加入20 mL乙腈和200µL内标溶液(1 mg/mL氘代蒽),超声萃取40 min,静置5 min,取4 mL上层清液于15 mL离心管中,加入3 mL正己烷-乙酸乙酯溶液(体积比3∶7),在涡漩振荡器上以500 r/min振荡5 min,静置,取上层清液净化。移取(1.5±0.2)mL上层清液于含有150 mg无水硫酸镁、50 mg PSA和50 mg C18吸附剂的2 mL离心管中,在涡漩振荡器上以500 r/min振荡5 min,再以5 000 r/min离心10 min,取上清液供气相色谱-质谱分析。
气相色谱-质谱条件:色谱柱为DB-35毛细管柱(30 m(长度)×0.25 mm(内径)×0.25µm(膜厚),固定相为含5%苯基的甲基聚硅氧烷)。进样口温度300℃。载气为氦气(纯度≥99.999%),恒流流速:1.0 mL/min。进样量1µL,分流进样,分流比40∶1。程序升温:初始温度70℃,以10℃/min升至300℃,保持5 min,后运行模式在300℃条件下保持5 min。传输线温度300℃;电离方式为电子轰击源(EI);电离能量70 eV;离子源温度280℃;四极杆温度150℃;溶剂延迟6 min。
1.2.2 微液节点采样-质谱成像离子源:正离子模式,喷雾针电压4 000 V,离子源温度200℃,雾化气(N2)流量1.5 L/min,干燥气压力100 kPa。全扫描模式:m/z100~1 000,离子累积时间40 ms;检测器电压1.59 kV。不同光引发剂质谱定性的选择离子质荷比及其对应的离子加合物分别为:EDB(m/z194.117 6,[M+H]+)、EHDBA(m/z278.211 5,[M+H]+)、MBF(m/z165.054 6,[M+H]+)、BDK(m/z257.117 2,[M+H]+)、OMBB(m/z241.085 9,[M+H]+)、PI1173(m/z165.091,[M+H]+)、BP(m/z183.080 4,[M+H]+)、2/3/4-MBP(m/z197.069 1,[M+H]+)、PI184(m/z205.122 3,[M+H+])、PI907(m/z280.136 6,[M+H]+)、2/4-ITX(m/z255.083 8,[M+H]+)、PBZ(m/z259.111 7,[M+H]+)、DETX(m/z269.099 5,[M+H]+)、MK(m/z269.164 8,[M+H]+)和DEAB(m/z325.227 4,[M+H]+)。
微液节点采样质谱装置见图1。微液节点探针由不同直径毛细管组成的同轴内外套管(外管外径365µm,内径250µm;内管外径150µm,内径100µm)。外管与PEEK三通的一端相连,而内管穿过三通通过套管和PEEK螺丝在三通的另一端固定。三通的第3个端口与注射泵连接用于给液。根据采样平台与质谱的两种接口,穿过三通一端的内管与质谱有两种接法(图1B-1和B-2)。两种成像方式的采样溶液均为90%乙腈-9.5%水-0.5%乙酸。成像方式1(图1B-1):采样流速调节控制5µL/min,定量环5.5µL,单点采样时间1 min,采样完毕后由液相泵用90%乙腈-9.5%水-0.5%乙酸溶液以0.1 mL/min的流速将定量环内的采样溶液推入质谱;成像方式2(图1B-2):采样流速不可调,主要由溶液组成以及质谱离子源喷雾提供的负压大小决定,本实验条件下流速稳定在5.5µL/min。采样溶液直接以此流速流入质谱离子源产生信号。成像方式1和2与质谱联用的场景照片分别见图1C-1和1C-2。
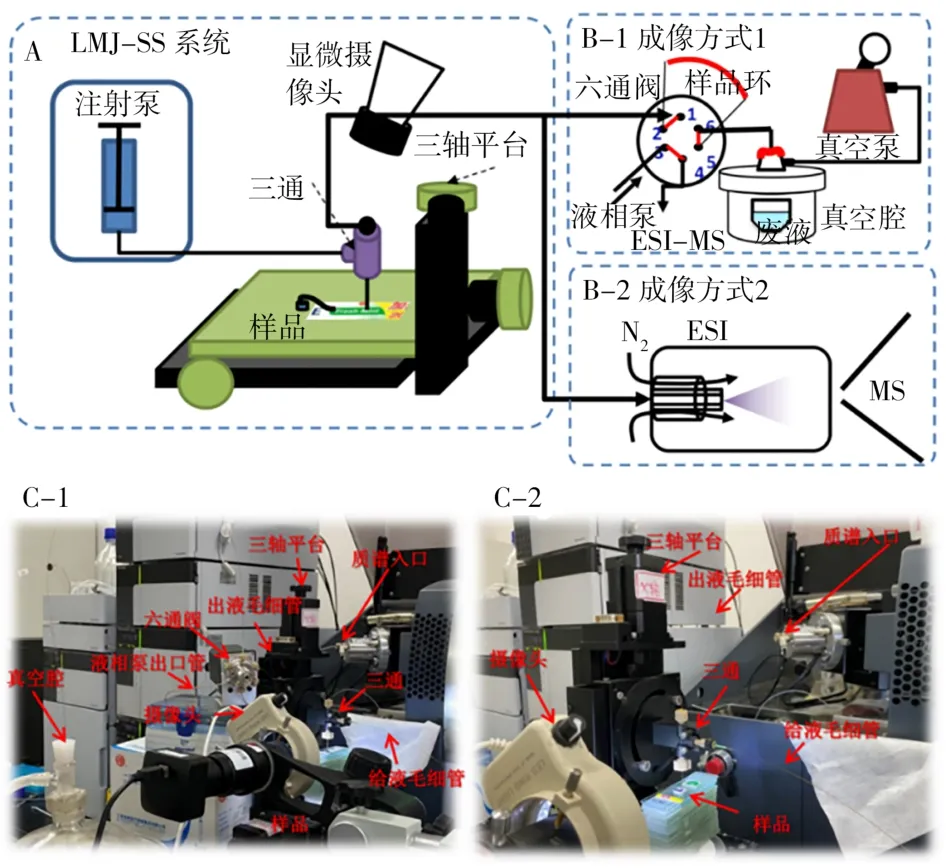
图1 微液节点采样质谱成像方式的示意图(A和B)与实物照片图(C)Fig.1 Schematic illustrations(A and B)and photos(C)showing the imaging mode of LMJSS-MS
微液节点探针固定在三轴电动平台上,由显微摄像头观察液节点的形成以及探针-样品之间的距离以及内外套管的距离,将探针与样品间的距离以及内外套管的距离均调节为50µm。成像时,由三轴平台控制探针在样品表面以200µm/s速度和400µm步间距逐点扫描,每个像素点停留1 min,同时采集每个像素点的质谱信号。
1.2.3 质谱成像的原位定量方法质谱可测得萃取液中分析物信号随时间的变化情况。分析物信号通过标准曲线(Sa=a*Ca+b)进行定量校正,从而得到分析物的浓度随时间的变化曲线,然后将每个像素点的浓度-时间曲线进行积分,得到采样时间内萃取的分析物总量(Mtotal):

式中Sa,i为单位采样时间上分析物的信号值,Δt为质谱采样时间间隔,u为采样流速。得到Mtotal后便可根据采样点的直径(rsampling)计算出分析物在纸张上的单位面积含量(Ctotal,mg/m2):

2 结果与讨论
2.1 微液节点采样以及质谱成像方式的选择
微液节点采样是一种基于原位液体萃取的具有空间分辨能力的采样方法。利用微液节点探针在样品表面进行扫描采集每个像素点的质谱图便可得到分析物在样品表面的质谱成像图。前期利用微液节点采样-质谱体系已经建立了包装纸中光引发剂的原位定量检测方法[13],该方法通过阀切换的方式实现稳定重复的样品采集和进样,虽然在纸张一定面积上扫描的方式可得到最大的检测灵敏度,但却降低了方法的空间分辨率,同时质谱成像的连续性也受到切阀过程复杂操作的影响。本研究将该方法改进后用于包装纸中光引发剂的定量成像。
目前微液节点采样方法用于质谱成像的接口技术主要有两种。第1种成像方式如图1B-1所示:萃取液由注射泵注入同轴套管的外管,而内管先与六通阀1位相连,六通阀6位再与隔膜泵的真空腔相连。通过隔膜泵提供的负压将液体由内管压入六通阀2-5位的定量环中进行收集。最后通过阀切换将2-5位的定量环与3-4位相连。由于3、4位分别连接液相泵和质谱离子源,所以阀切换后定量环中收集的样品被注射进入质谱检测。这种方式与前期方法[13]的装置结构一致,只是将探针在一定面积内(约12 mm2)的扫描变为单个采样点(0.12 mm2)的萃取,以提高采样的空间分辨率。第2种成像方式如图1B-2所示,萃取液由注射泵注入同轴套管的外管,而内管与电喷雾喷针相连,通过电喷雾产生的负压将液体由内管压入质谱离子源,在探针尖端形成稳定的液节点。
由此可见,成像方式2更加简单,可实时得到微液节点采样的质谱信号曲线,对萃取过程进行监测。但其负压依靠质谱离子源喷雾产生,采样的溶剂流速难以调节,控制性略差。成像方式1装置复杂,但其隔膜泵的压力可调,整个采样过程可调节的参数更多,稳定性更好,适用范围更宽。由于两种方法各有优势,需要将其进行对比检测。由于前期研究已获得光引发剂萃取的最优溶剂为90%乙腈-9.5%水-0.5%乙酸,因此利用该溶剂比较两种采样方式对实际包装纸中光引发剂检测的灵敏度和重复性(图2)。由图可知,成像方式1的采样信号远低于成像方式2,对于同一包装纸成像方式1仅检出4种光引发剂,而成像方式2可检出7种光引发剂。其原因主要为:①成像方式1得到的是将萃取液收集到定量环后进样的平均质谱信号,对萃取液有稀释作用,无法得到萃取动力学曲线中最高浓度的信号值;②成像方式1收集在定量环中的萃取液需经液相泵中的溶剂推入质谱,这时萃取液又会被稀释,进一步降低了进样浓度。由此造成成像方式1的灵敏度低。此外,图2中也可看出成像方式1的检测重复性略优于成像方法2,这与其体系的稳定性以及平均了一段时间的采样信号有关。成像方式2得到萃取分析物信号相对于时间的曲线,其中包含了采样过程的信号波动和萃取效率波动,综合两种方式灵敏度和重复性,选择灵敏度更高而重复性略差的成像方式2。另外,成像方式2相较于先前开发的原位定量检测方法[13]在提供同样空间分辨率的前提下具有10倍以上灵敏度的提高,只是重复性略有降低。

图2 两种采样方式检测光引发剂的结果对比Fig.2 Comparison of two sampling methods for detection of photoinitiators
2.2 微液节点采样动力学曲线
成像方式2可得每个像素点采样的动力学曲线(即信号-时间曲线),由曲线可得微液节点在不同包装纸位置的采样速率差别。如图3所示,选取包装纸上不同油墨颜色(红色、绿色和蓝色)的几个位置分别进行微液节点采样,采样时间均在1 min以上。由结果可见,不同位置光引发剂信号随时间的变化都是先上升后下降的过程,这服从异相传质的动力学理论[15-16]。由前人的推导[15]可知,分析物在两相间传质的扩散层一旦建立后,单位时间内分析物的传质量主要由两相之间的浓度差以及扩散层厚度决定。由于扩散层厚度在传质过程中基本不变,所以传质量主要由浓度差决定。微液节点采样的萃取液是不断更新的,所以液相中的分析物浓度可看作实时更新,一直为0,而固相样品中的分析物浓度则不断减少,因此,在传质过程中随着浓度差不断减小,单位时间的传质量也不断减小,采样后期分析物信号不断下降。本课题组在近期研究中推导了单点采样过程中萃取液中的分析物浓度随采样时间的变化曲线[17],由推导公式可知,萃取液中分析物的浓度随时间呈指数级下降,而下降速度主要由时间常数决定。在模型中时间常数主要受传质系数和传质层厚度影响,传质系数越大、传质层厚度越薄则时间常数越大,从而下降速度越快。而在峰值之前的信号上升过程主要是扩散层未完全建立,传质面积不断扩大产生的,而且这个过程通常时间较短。

图3 不同包装纸位置上进行微液节点采样的信号-时间曲线Fig.3 Signal-time curves of LMJSS at different positions of packing paper
对比3个不同位置的信号-时间曲线(图3A~C),发现虽然不同位置其曲线上升与下降的速度是不同的,但基本上分析物信号最终都降至0附近。由于最终信号基本降为0,可由1 min内分析物信号随时间的积分面积除以信号几近为0的时间内的信号积分面积计算得采样1 min时的萃取回收率(考虑到在同一样品点的采样过程中萃取液中的基质效应基本不变,分析物的离子化效率也基本不变,所以可用信号值代替分析物在萃取液中的浓度值进行计算)。结果显示,在采样1 min时,体系对不同包装纸位置都能完成单个像素点内分析物90%的萃取。由此可知,当单点萃取时间为1 min时,不同位置的萃取效率基本一致,不存在由不同位置萃取效率差别而引起的空间分布以及相对定量检测的误差。这种方法无需通过样品表面加标即可对纸张中的分析物进行绝对定量。因此,后期在进行成像时选择1 min为每个像素点的采样时间,通过对采样时间内的信号-时间曲线进行积分得到每个点的分析物信号值。
对于定量成像而言,除了不同位置萃取效率的差别外,不同位置的基质效应导致的离子抑制或增强同样会影响定量,通常采用在萃取液中加入内标来消除和校正。而本课题组的研究已证明选取的最优萃取溶剂可采用外标校正法,且基质效应很小[13]。具体操作为将系列浓度的标样溶于萃取液中利用与实际微液节点采样同样的流速和条件进样于质谱中,标样的质谱信号用于校正实际信号得到分析物在萃取液中的绝对浓度。由图3的讨论可知,分析物在不同包装纸位置均能实现90%以上的回收率,所以计算得到萃取液中的待测物浓度可以换算成分析物在一个像素点采样面积的纸中的含量,用含量除以纸张的采样面积即可得到分析物在纸中的绝对浓度(计算方法见“1.2.3”)。
2.3 方法在包装纸中光引发剂成像上的应用
2016年,中国环境保护部科技标准司发布了HJ/T 2524-2016《环境标志产品技术要求胶印油墨》,该标准明确提出能量固化油墨不添加二苯甲酮(BP)、异丙基硫杂蒽酮(ITX)、2-甲基-1-(4-甲硫基苯基)-2-吗啉基-1-丙酮(907)作为光引发剂[3],EHDBA作为硫杂蒽酮类光引发剂ITX的协同试剂,主要增强ITX在油墨中的固化作用[4],所以ITX和EHDBA的检测均很重要。2011年,德国宣布召回从比利时进口的冷冻细面条,主要原因是面条包装上印刷油墨所含有的二苯甲酮(BP)渗入面条中,导致面条被污染,迁移量达1 747µg/kg[18]。由此可见包装纸中BP检测以及油墨中光引发剂溯源的重要性。
将建立的微液节点采样-质谱成像方法应用于包装纸中多种光引发剂的定量成像(图4A)。选取涵盖包装纸5种颜色的3个区域进行扫描成像,分别是涵盖红色、白色、黄色油墨的区域;涵盖绿色、白色油墨的区域;以及涵盖蓝色、白色油墨的区域。定量成像结果如图4B的热图所示,不同区域能检测到光引发剂的种类和含量都有很大差别。而在一个区域内,光引发剂的分布也各不相同。将3个区域对比发现,黄红色区域检出的光引发剂种类最多,包括PI907、ITX、PBZ、DETX、DEAB、EDB、EHDBA和OMBB,含量在0~5 mg/m2之间,且部分光引发剂的分布与油墨的图形相吻合。如ITX、PBZ、EHDBA和OMBB 4种光引发剂均集中分布于红色油墨图形的区域,而在黄色部分的含量很少。PBZ和EHDBA的分布尤其明显,只分布在红色油墨区域,黄色区域几近于0。特别是EHDBA,其含量较低(约1 mg/m2),分布的集中性会使得传统大面积裁样的定量方法难以检出。本实验利用质谱成像的空间分辨能力提高了ITX和EHDBA的检测灵敏度以及空间分布信息,这对于ITX和EHDBA两种禁用光引发剂的筛查具有重要意义。而绿白色区域检出的光引发剂与红黄色区域差别很大,主要包括PBZ、DETX、EDB、EHDBA、OMBB、BP和PI184;光引发剂在绿色和白色位置的分布差别很小,看不出明显与油墨图形对应的分布,说明这两种颜色的油墨采用的光引发剂基本相同。绿白色区域与其他两个区域相比,BP和PI184的含量(约5 mg/m2)显著高于其他两个区域,这为高含量光引发剂的溯源提供了重要信息。在蓝白区域,可检出的光引发剂包括PI907、ITX、PBZ、DETX、DEAB、EDB和EHDBA。这些光引发剂的含量基本与红黄区域一致,但主要分布在蓝色油墨位置,而在白色位置含量很低。
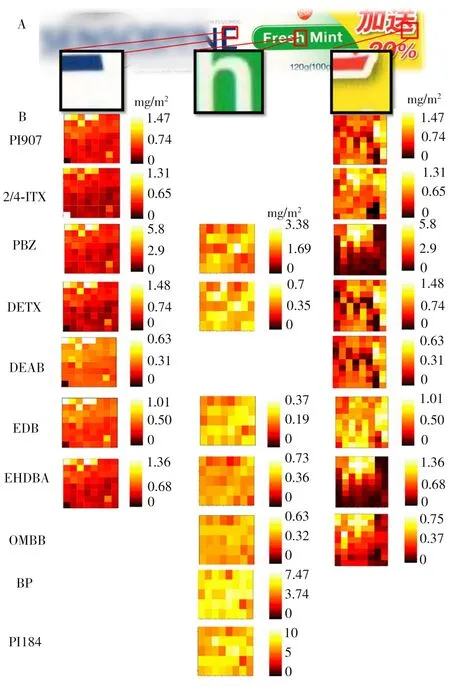
图4 采用微液节点采样-质谱系统对包装纸中不同区域(A)的光引发剂进行成像的定量成像图(B)Fig.4 Quantitative imaging of PIs in different areas(A)of packaging paper by using LMJSS-MS system(B)
从成像的结果可见,有些光引发剂只在特殊颜色或图案处才可检出,这是由于包装纸通常采用多种印刷工艺制造而成。有些印刷工艺如丝印技术或胶印技术只在包装纸的很小区域内使用,利用传统的采样、前处理和检测方法进行处理和测试时,仅能得到包装纸主区域中光引发剂的平均含量信息,可见质谱成像可提供传统定量方法无法提供的光引发剂空间分辨信息,对光引发剂成分的溯源优势明显。
2.4 成像结果与标准质谱定量方法结果的对比
为了验证成像结果的准确性,将同样的包装纸取图5A所示的红框位置进行传统标准方法(气相色谱-质谱)的定量,前处理及检测方法见“1.2.1”,结果如图5B红柱所示。成像定量结果通过不同颜色区域在标准方法采样区域对应的面积换算的定量值由图5B黑柱所示。结果显示,传统方法与成像方法的定量结果在一个数量级范围内,但稍有偏差。如定量结果相差比较大的BP,由于其在使用质谱检测时信号很低,因此信号-时间曲线中难以判断其绝对的采样回收率,同时也造成其检测误差较大。另外还有一些光引发剂只能在一种方法中检出,如传统方法中检出的MBP利用成像方法无法检出,这是因为传统方法采用气相色谱-电子轰击离子源-质谱检测,而成像方法采用电喷雾离子源质谱检测,两种电离方式的电离范围不同,电喷雾电离源难以检测MBP这种低极性化合物。而成像方法检出的3种光引发剂ITX、EDB和EHDBA利用传统方法无法检出。由此可见,质谱成像对于空间分布集中的一些光引发剂具有提高灵敏度的作用,传统定量方法采用大面积采样的方式,其对分布集中的光引发剂造成很大的稀释,以至于无法检出。本文建立的成像方法对这些禁用或限用光引发剂的筛查和检测具有重要意义。

图5 传统GC-MS定量方法的取样区域(A)、其与定量成像方法的结果比较(B)以及定量成像方法得到光引发剂在不同油墨区域的定量比例(C)Fig.5 The sampling area of the method of GC-MS(A),the comparison of the results with two methods(B)and the ratio of PIs in different ink areas obtained by the imaging method(C)
图5C展示的是成像结果得到的红框采样区域3种不同颜色的油墨所对应的光引发剂含量占比。由图可见,不同光引发剂在不同油墨区域中的占比有很大差别,而这个差别只能通过有空间分辨能力的成像技术才能显示出来。
3 结 论
本研究利用微液节点采样-质谱系统建立了包装纸中光引发剂的定量成像方法,并将其用于包装纸中多种光引发剂的检测。结果表明,成像方法定量结果与传统方法基本一致,且可以检测到种类更多的光引发剂。另外,成像方法的空间分辨率还可提供传统方法无法得到的光引发剂在包装纸的空间分布信息,这对于禁用或限用光引发剂的溯源和检测是非常有用的。另外,此方法对于指导和改进光固化油墨的涂布配方或工艺具有重大意义。
猜你喜欢
绿色包装(2022年9期)2022-10-12
现代仪器与医疗(2022年1期)2022-04-19
现代仪器与医疗(2021年2期)2021-07-21
孩子·小学版(2021年5期)2021-06-04
中国科技教育(2020年3期)2020-10-21
分析化学(2019年3期)2019-03-30
分析化学(2018年12期)2018-01-22
红蜻蜓(2016年6期)2016-05-14
红蜻蜓(2014年2期)2014-02-19
