
Artificial Intelligence Cracks a 50-Year-Old Grand Challenge in Biology
2021-11-26 03:46:22SeanNeill
Engineering 2021年6期
Sean O’Neill
Senior Technology Writer
In late November 2020, DeepMind Technologies, the Londonbased, artificial intelligence (AI)-focused subsidiary of Google’s parent company, Alphabet, announced that its AlphaFold system had achieved ‘‘unparalleled levels of accuracy” in predicting the complex shape of proteins based solely on their genetic sequences[1]. The feat meets a 50-year-old grand challenge in biology, the extraordinarily difficult problem of predicting how proteins fold.The advance is expected to have a significant impact on drug discovery and the burgeoning field of protein design, possibly even helping to tackle the coronavirus disease 2019 (COVID-19) pandemic[2],especially with the rapid emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants [3].
‘‘Protein folding is one of these holy grail-type problems in biology,” said Demis Hassabis, founder and chief executive officer of DeepMind, at the time. ‘‘We have always hypothesised that AI should be helpful to make these kinds of big scientific breakthroughs more quickly.”
Proteins are large,complex molecules that play a key role in virtually every aspect of the biological world.It is the shape of proteins that define their functions: hemoglobin transports nutrients,enzymes catalyse chemical reactions, collagen provides structure,insulin regulates blood glucose, and antibodies provide immunity.These and all other proteins are created from the same palette of 20 amino acids in the standard genetic code, connected in long chains.
Constructed amino acid by amino acid by living organisms or through synthetic processes, proteins naturally twist and fold together into complex shapes, full of bends, helixes, and sheets.Antibody proteins are ‘‘Y”-shaped, for example, which enables them to latch on to and help neutralize disease-causing bacteria or viruses. Conversely, harmful genetic mutations can lead to the production of misfolded, non-functional proteins, such as those that cause cystic fibrosis.
The code for producing proteins is contained in deoxyribonucleic acid (DNA). But while DNA sequencing reveals the sequence of amino acids that a given protein comprises,it does not tell how they fold into their ultimate shape.And the larger a protein’s sequence,the more difficult it becomes to predict its shape.The chain of a typical protein could,in theory,fold into any of an astronomical number of conformations, making attempts at brute force calculation futile[4].
The protein folding challenge originated in 1972 when, in his acceptance of the Nobel Prize in Chemistry, the American biochemist Christian Anfinsen declared that the amino acid sequence of a protein should be sufficient to determine,in a specific environment, its folded shape [5]. For decades, however, the only way to accurately determine the shape of a protein of interest has been to use expensive and painstaking methods such as nuclear magnetic resonance and X-ray crystallography,and,more recently,cryo-electron microscopy. It can take years of such experimental work to delineate the shape of a single protein,with no guarantee of success.
In 1994, in a bid to coalesce a global community of scientists around the problem, John Moult, a professor of cell biology and molecular genetics at the University of Maryland in Rockville,MD,USA,and colleagues created a large-scale experiment to assess computational methods for generating protein structures [6]. This effort became the biennial Critical Assessment of Structure Prediction (CASP) event, which Hassabis refers to as the ‘‘Olympics of protein folding.”
The CASP competition has three rolling stages: ①collecting about 100 protein targets, the shapes of which have recently been uncovered by lab work,but crucially,not yet published;②providing the genetic sequences of these targets to teams around the world, which then set to work using software systems to predict their shapes; and ③blindly assessing the submitted predictions.CASP judges the accuracy of the predicted shapes primarily using a measure called the ‘‘Global Distance Test” (GDT), which ranges from 0 to 100. Moult said that a score of around 90 is comparable to results obtained through experimentation.
Progress since 1994 had been steady but slow—until CASP13 in 2018,when DeepMind entered for the first time,with an early version of AlphaFold[7].The team won by a large margin,startling the CASP community, but AlphaFold’s predictions were still far from the actual structures of the target proteins, with a median GDT of 59 (Fig. 1).
For CASP14 in 2020, however, DeepMind came back with a completely revamped AlphaFold, and this time the results were stunning.‘‘It was extraordinary,”said Moult.‘‘You see one surprising prediction come in, and you think, ‘what’s going on here?’. By when you have three or four structure predictions that are unbelievably accurate, you realise something very important has happened.”
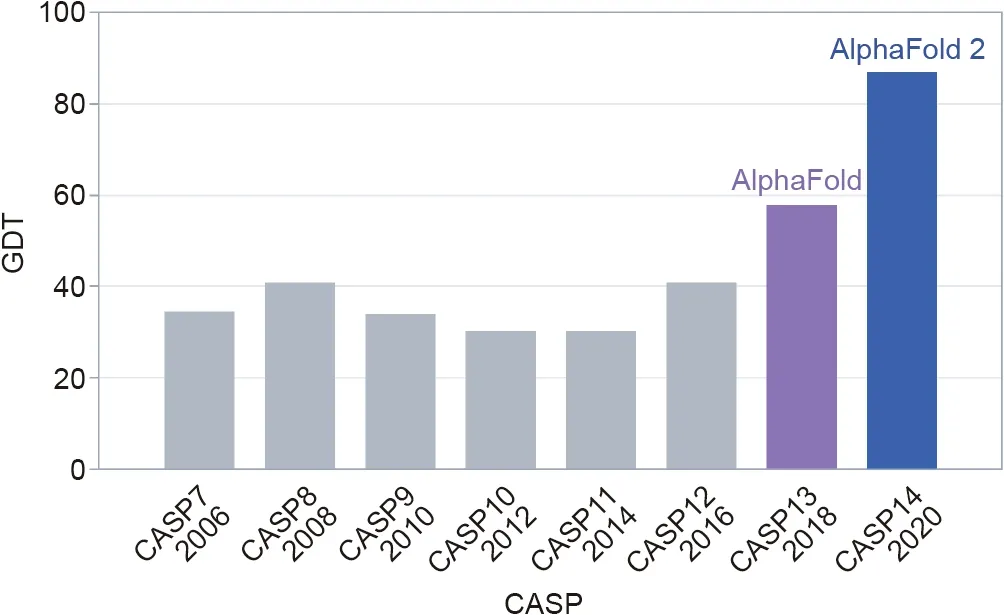
Fig. 1. The median accuracy of the winning team’s predictions—using a measure called the GDT—in the free-modelling category, the toughest category in the biennial CASP event.DeepMind’s AlphaFold system took first place in both the 2018 and 2020 competition. Credit: DeepMind, with permission.
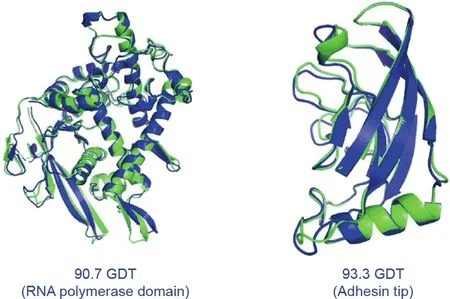
Fig. 2. The structures of several proteins predicted as part of CASP14 by AlphaFold(blue) superimposed on experimentally determined structures (green). They are remarkably close matches. RNA: ribonucleic acid. Credit: DeepMind, with permission.
AlphaFold scored 87 GDT in the hardest category,with a median score of 92.4 GDT across all the protein targets(Fig.2)[8].The system’s average error is approximately 0.16 nanometres—roughly the width of an atom. To deliver this coup, the DeepMind team developed a novel, attention-based neural network system [9]. In machine learning, ‘‘attention” means a design that mimics human attention, insofar as the system identifies key aspects of the data and gives those more weight,while paying less attention to aspects of the data that it deems less important.In-depth technical details of this deep-learning system are yet to be shared—but peerreviewed papers are expected later this year. AlphaFold (Fig. 3)[1]was trained using publicly available data from the Protein Data Bank (PDB)—which contains the structures of about 175 000 proteins—in addition to other large databases containing the sequences of proteins of unknown structure. The training period required 16 or so Google TPUv3 coprocessors (equivalent to between 100–200 graphic processing units) run over ‘‘a few weeks,” according to the DeepMind team, with individual protein structure predictions completed ‘‘in a matter of days” [1].
Moult has heard neural networks dismissed as glorified pattern recognition, yet the degree of atomic-level knowledge that Alpha-Fold was able to distill from its training was remarkable, he said.‘‘The level of abstraction it achieved was profound. It is as if the machine, in an alien sense, has learned the physics. It can take any situation in which protein-type structures are involved and get it right at the atomic level.You cannot do that just by recognizing a set of patterns in the training data.”
The breakthrough opens opportunities across biology, but drug discovery is where it may have its most immediate impact. Most drugs work by binding to proteins in the body, triggering changes in how they function. With machine-learning systems like Alpha-Fold, it should become possible to quickly work out the shape of proteins of interest, and then design drugs—or repurpose existing ones—to bind effectively to those proteins.
For example, as the scale of the coronavirus pandemic became evident in early 2020, and later as part of CASP14,DeepMind took the genetic sequences of several proteins that form part of the SARS-CoV-2 virus and provided structural predictions that were then largely borne out by experiment [10]. Such work has the potential to speed up the design of drugs that could counteract the disease. In fact, protein design is the flip side of shape prediction: Once a machine has a firm understanding of the atomic processes that underpin protein folding, it becomes easier to design proteins that fold into the shape required.
‘‘We’ve been using current protein design methods to develop COVID-19 therapeutics, vaccines, and sensors that look very promising and are already in, or headed for, clinical trials,” said David Baker, director of the Institute for Protein Design, based at the University of Washington in Seattle,WA,USA,who led the team that came in second to DeepMind at CASP14[11].‘‘With improved protein design,we should be able to do even better,faster.”
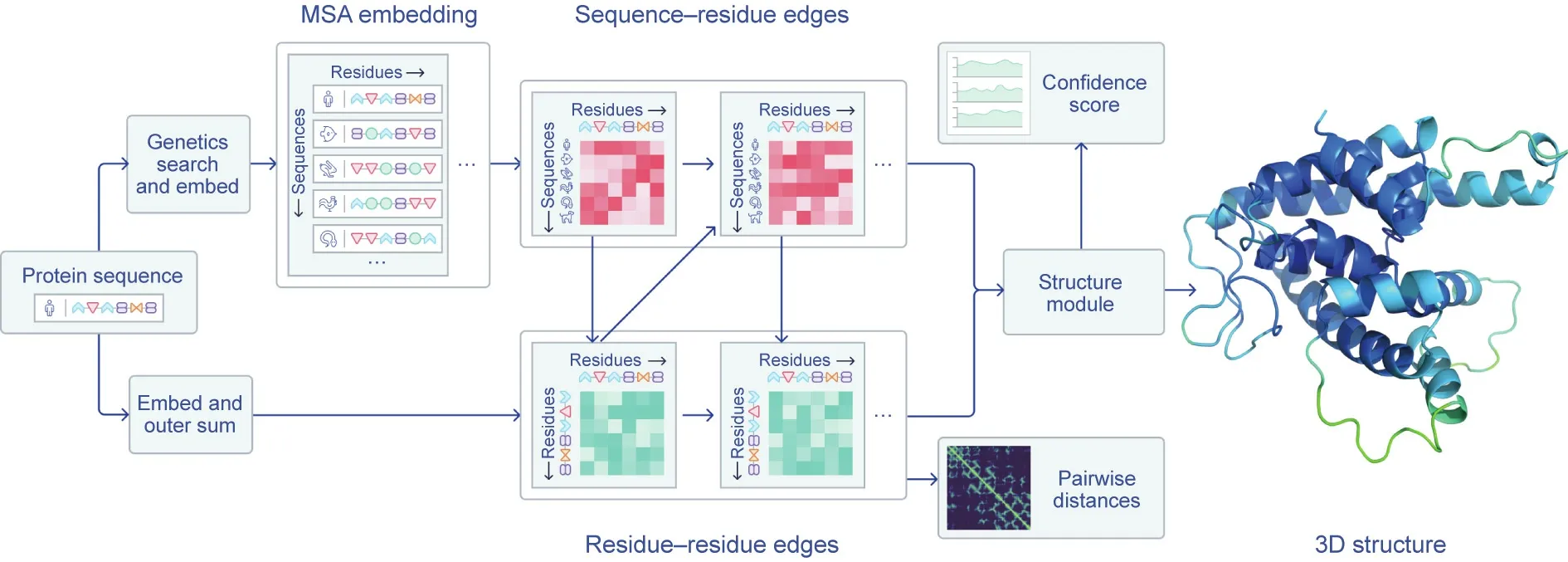
Fig. 3. An overview of AlphaFold’s architecture. DeepMind has yet to provide in-depth details about its system but describes how ‘‘a folded protein can be thought of as a‘spatial graph,’where amino acid residues are the nodes and edges connect the residues in close proximity”[1].MSA:multiple sequence alignment;3D:three-dimensional.Credit: DeepMind, with permission.
Technology like AlphaFold could also be used to explore proteins and enzymes that might be used to break down industrial waste, or old plastics, for example, or efficiently draw carbon out of the atmosphere. ‘‘The immediate impact on the field of structural biology is huge,”said Osnat Herzberg,a professor of biochemistry at the University of Maryland and contributor of protein structures to CASP14. ‘‘These approaches will have important medical applications and lead to technological advances that we currently cannot imagine.”
A more cautious note was sounded by David Jones,professor of bioinformatics and head of the Bioinformatics Group at University College London.‘‘Results like this have woken people up to the fact that machine learning can have a huge influence beyond the obvious areas of machine vision and natural language processing,”Jones said. ‘‘But I am not amongst the people who believe we will have new treatments for diseases just because we can now model protein structures much more accurately than we could before.It is important to test systems as complex as this under a lot of different conditions before we can be sure of what its capabilities or limitations are.”

- Engineering的其它文章
- A Novel Attribute-Based Encryption Approach with Integrity Verification for CAD Assembly Models
- Gaze Estimation via a Differential Eyes’ Appearances Network with a Reference Grid
- Editorial for the Special Issue on Intelligent Manufacturing
- Coronavirus Variants—Will New mRNA Vaccines Meet the Challenge?
- The State-of-the-Art Review on Applications of Intrusive Sensing, Image Processing Techniques, and Machine Learning Methods in Pavement Monitoring and Analysis
- Bioprinting of Small-Diameter Blood Vessels