
Regulatory role of the transforming growth factor-β signaling pathway in the drug resistance of gastrointestinal cancers
2021-11-20 06:33:10XiaoqunLvGuoxiongXu
Xiaoqun Lv,Guoxiong Xu
Xiaoqun Lv,Department of Pharmacy,Jinshan Hospital,Fudan University,Shanghai 201508,China
Guoxiong Xu,Research Center for Clinical Medicine,Jinshan Hospital,Fudan University,Shanghai 201508,China
Guoxiong Xu,Department of Oncology,Shanghai Medical College,Fudan University,Shanghai 200032,China
Abstract Gastrointestinal(GI)cancer,including esophageal,gastric,and colorectal cancer,is one of the most prevalent types of malignant carcinoma and the leading cause of cancer-related deaths.Despite significant advances in therapeutic strategies for GI cancers in recent decades,drug resistance with various mechanisms remains the prevailing cause of therapy failure in GI cancers.Accumulating evidence has demonstrated that the transforming growth factor(TGF)-β signaling pathway has crucial,complex roles in many cellular functions related to drug resistance.This review summarizes current knowledge regarding the role of the TGF-β signaling pathway in the resistance of GI cancers to conventional chemotherapy,targeted therapy,immunotherapy,and traditional medicine.Various processes,including epithelial-mesenchymal transition,cancer stem cell development,tumor microenvironment alteration,and microRNA biogenesis,are proposed as the main mechanisms of TGF-β-mediated drug resistance in GI cancers.Several studies have already indicated the benefit of combining antitumor drugs with agents that suppress the TGF-β signaling pathway,but this approach needs to be verified in additional clinical studies.Moreover,the identification of potential biological markers that can be used to predict the response to TGF-β signaling pathway inhibitors during anticancer treatments will have important clinical implications in the future.
Key Words:Drug resistance;Gastrointestinal cancer;Transforming growth factor-β;Epithelial-mesenchymal transition;Cancer stem cells;MicroRNAs
INTRODUCTION
Gastrointestinal(GI)cancer,including esophageal cancer(EC),gastric cancer(GC),and colorectal cancer(CRC),is one of the most prevalent types of malignant carcinoma,falling within the top six in mortality according to global cancer statistics in 2018.In both sexes,CRC is the second leading cause of cancer death(9.2% of total cancer deaths),closely followed by GC(8.2%),and EC as the sixth leading cause of mortality(5.3%)[1].CRC is also the second most common cause of cancer death in the United States[2].Despite improvements in current therapeutic strategies,including surgery,radiotherapy,chemotherapy,targeted therapy,and immunotherapy,clinical prognoses and therapeutic responses of GI cancer patients are far from satisfactory because of delayed diagnosis,recurrence,poor clinical response,high cost,and medication side effects[3,4].
Chemotherapy is the most commonly used treatment for patients with advanced GI cancer.The most widely used chemotherapeutic regimens for GI cancer are fluorouracil and platinum[5-7].Despite the continual development of new chemotherapeutic strategies,resistance to anticancer drugs remains a significant problem that is responsible for unfavorable clinical outcomes and treatment failures.Chemoresistance,including intrinsic and acquired drug resistance,is defined as the resistance of cancer cells to various structurally and functionally unrelated anti-cancer drugs[8].The mechanisms of drug resistance are complex and closely related to various signaling pathways that are activated by many stimuli to promote chemoresistance[9].
The transforming growth factor(TGF)-β signaling pathway is deregulated in cancer and can have tumor-suppressive or tumor-promoting roles,depending on the molecular and cellular context[10,11].In the GI tract,TGF-β has crucial and complex roles in many cellular functions related to drug resistance,such as maintaining stem cell homeostasis,regulating epithelial to mesenchymal transition,modulating immunity,and promoting fibrosis[12,13].In this review,we discuss the role of the TGF-β signaling pathway in regulating chemoresistance in GI cancers.
MECHANISMS OF CHEMORESISTANCE IN CANCER
Molecular investigations have revealed several mechanisms underlying chemoresistance,including the epithelial-mesenchymal transition(EMT),the efflux of intracellular chemotherapeutic drugs,noncoding RNAs,stem cell development,and the tumor microenvironment[14-17].EMT is a complex and important cellular program in which epithelial cells shed their differentiated characteristics and acquire mesenchymal phenotypes,including motility,invasiveness,and resistance to apoptosis.Cells undergoing EMT become more invasive and exhibit increased resistance to anticancer drugs[18,19].In addition,EMT has been found to result in stem cell-like characteristics and is positively correlated with the expression of ATPbinding cassette(ABC)transporters[18,20,21].Different stimulus-induced EMT may contribute to chemoresistanceviathe upregulation of distinct transcription factors[19].
Failure of cancer chemotherapy can also be caused by changes in the expression or activity of membrane transporters,primarily those belonging to the ABC transporter family.ABC transporters can export chemotherapeutic agents out of the cell,thereby reducing intracellular drug levels and drug sensitivity and ultimately contributing to cancer chemoresistance[22,23].In addition,ABC proteins transport signaling molecules that contribute to tumorigenesis[24].
Increasing evidence shows that non-coding RNAs,especially microRNAs(miRNAs)and long non-coding RNAs(lncRNAs),can affect chemoresistance by forming a competing endogenous RNA regulatory network with mRNAs[25].MiRNAs can play roles in drug resistance by targeting hundreds of tumor-related gene transcripts and affecting complex molecular pathways[14,26].Specific miRNAs may be used as potential predictive biomarkers to guide individualized chemotherapy by reversing drug resistance[14].
Cancer stem cells(CSCs),which make up a distinct population within the tumor mass,possess unique self-renewal,multilineage differentiation,and potent tumorigenic abilities[27,28].These cells acquire chemoresistance through various pathways involving apoptosis and DNA repair mechanisms[29].In addition,upon exposure to cytotoxic therapies,CSCs can convert non-CSCs to CSC-like cells that may persist after treatment and serve as a mechanism for relapse.In GI malignancies,CSCs are abundant and contribute to chemotherapeutic resistance[15].
Interactions of tumor cells with alterations of the microenvironment,such as energy deprivation,hypoxia,and inflammation,give rise to heterogeneity and chemoresistance.Most tumor cells display deviations from the normal energy metabolism,allowing them to survive in hypoxic and low nutrient microenvironments[30,31].Mitochondrial dysfunction and fatty acid(FA)metabolism are associated with chemotherapeutic resistance[31,32].Hypoxia can also drive tumor resistance to chemotherapy by upregulating hypoxia-inducible factor-1(HIF-1)and its downstream genes[33].Inflammation and inflammatory mediators,including TGF-β,have been shown to contribute to the development,progression,metastasis,and chemoresistance of cancer[34,35].In addition,the gut microbiota,which is linked to chronic inflammation and carcinogenesis[36],has an important role in the modulation of the host response to antitumor treatments,especially chemotherapy and immunotherapy[37].Moreover,emerging evidence has demonstrated that cancer-associated fibroblasts(CAFs),one of the critical components of the tumor microenvironment,confer substantial resistance to chemotherapy and influence tumor cell responsiveness to immune checkpoint inhibitors[38].
ROLE AND ALTERATIONS OF THE TGF-β SIGNALING PATHWAY IN GI CANCER
The TGF-β signaling pathway can be subdivided into canonical Smad-dependent and noncanonical Smad-independent pathways.In the canonical pathway,TGF-β initially binds to the TGF-β type 2 receptor(TβRII),which recruits and phosphorylates the kinase domain of TGF-β type 1 receptor(TβRI),leading to the activation and phosphorylation of Smad2 and Smad3.Then,phosphorylated Smad2 and Smad3 bind to Smad4,allowing the entire complex to translocate into the nucleus.In the nucleus,the Smad complex regulates transcriptional activity by interacting with Smad binding elements within downstream target genes[39-41].Smad7 negatively regulates the TGFβ signaling pathway by blocking the interaction between Smads and receptors and inhibiting the phosphorylation of Smad2 and Smad3[42,43].In addition to the Smaddependent pathway,the binding of the TGF-β ligand to its receptors also activates several Smad-independent signaling pathways,including the mitogen-activated protein kinase,phosphoinositide 3-kinase(PI3K)/AKT,and Rho-associated protein kinase pathways[44,45].
The TGF-β signaling pathway has an important role in controlling tissue development,proliferation,apoptosis,differentiation,and homeostasis[46].Disruption of this signaling pathway leads to various diseases,including some cancers.In cancer cells,TGF-β signaling causes EMT and CSC-like traits,resulting in an aggressive phenotype and a poor prognosis[47,48].In addition to its direct effect on epithelial tumor cells,TGF-β controls tumor development by regulating the tumor microenvironment and growth factors from the surrounding stroma[13,49].Furthermore,TGF-β signaling activation in the tumor microenvironment suppresses antitumor immune responses and supports cancer cell survival[50].TGF-β has been found to inhibit multiple components of the immune system,including natural killer cells,CD8+cytotoxic T lymphocytes,B-cell proliferation,and immunoglobulin A secretion[51].Therefore,the TGF-β signaling pathway is associated with drug resistance and immune system escape.
In CRC,TGF-β1 expression is markedly increased and is correlated with poor clinical outcomes and a high risk of relapse[52,53].TGF-β1 expression is also increased in GC mucosa and precancerous gastric cells[54,55].However,active TGF-β1 is expressed most highly in smooth muscle actin-positive fibroblasts rather than in the malignant epithelial cells of gastric tumors[56].In GC patients,high serum and tissue TGF-β1 levels are associated with lymph node involvement and poor prognosis[57].Moreover,increased expression of TGF-β is found in EC[58].In sum,serum and tissue TGF-β levels are upregulated in GI cancers and are associated with metastases and poor prognoses.Alterations in the TGF-β signaling pathway,especially receptor andSmadgene mutations,are commonly observed in GI cancers where they lead to tumor formation and metastasis[13].Mutations in the TGF-β signaling pathway are found in 80% of CRC cell lines and approximately one-third of CRC tumors[46].A decreased or complete loss of TGF-β receptor expression is common in patients with esophageal adenocarcinoma,primary gastric tumors,and CRC[49].TβRII mutations frequently occur in the advanced stages of the colon[59]and gastric tumors along with progressive microsatellite instability(MSI-H)[49,60].The overall incidence of TβRII mutations is approximately 30% in CRC,while frameshift mutations can be found in approximately 80% of MSI-H CRC[60,61].TβRII mutations in CRC cells can contribute to the malignant phenotypeviamultiple pathways,regulate the components secreted by cancer cells,and directly promote inflammation in the tumor microenvironment[50].Compared with TβRII,mutations in its counterpart TβRI are less frequent in both CRC and GC[13,60].
A study of over 700 cases of sporadic CRCs reveals that the prevalence of Smad4,Smad2,and Smad3 mutations was 8.6%,3.4%,and 4.3%,respectively,with a combined prevalence of 14.8%[62].Both Smad2 and Smad4 are located on chromosome 18q,which is commonly deleted in CRC[63].However,Smad2 and Smad4 mutations tend to occur in the early and advanced stages of CRC,respectively[13,61,64].Loss of Smad4 contributes to colorectal carcinogenesis[46]and may be a predictive biomarker of the response to 5-fluorouracil(5-FU)-based chemotherapy[65].In GC,the expression of Smad3 is low or even undetectable in 40% of tissues,so mutations in Smad2 and Smad3 have not been described[13].
TGF-β SIGNALING AND DRUG RESISTANCE IN GI CANCER
Accumulating evidence suggests that the expression levels of components of the TGFβ signaling pathway are closely associated with response to chemotherapy.Immunohistochemical analysis of 78 patient biopsies reveals that p-Smad2/3 expression was elevated in C-type CRC tumors,which benefit the least from chemotherapy[66].Mediator Complex Subunit 12(MED12)negatively regulates TβRII through physical interactions;therefore,its suppression induces the activation of TGF-β signaling[67].In CRC cells,both MED12 knockdown and recombinant TGF-β treatment result in resistance to cisplatin,oxaliplatin(OXA),and 5-FU[66,67].However,another study shows that TGF-β2 suppression was associated with recurrence in patients with colorectal adenocarcinomas.In addition,disease-free survival(DFS)and overall survival(OS)are significantly longer in patients with tumors expressing TGF-β2[68].Additionally,in esophageal squamous cell carcinoma(ESCC)patients,TGF-β 1–509C/T polymorphisms benefit from radiochemotherapy and therefore might be useful genetic markers for predicting radiochemotherapy response[69].In GI cancers,the TGF-β pathway is correlated with resistance to antitumor agents,including conventional chemotherapy,targeted therapy,immunotherapy,and traditional medicine.In Table 1,we provide a summary of the relationships between the TGF-β signaling pathway and drug resistance in GI cancers.

Table 1 Studies of the transforming growth factor-β signaling pathway in drug resistance in gastrointestinal cancer

5-FU:5-fluorouracil;BETi:Bromodomain and extraterminal domain protein inhibitors;BMP-4:Bone morphogenetic protein 4;CRC:Colorectal cancer;DDP:Cisplatin;Dox:Doxorubicin;EC:Esophageal cancer;ESCC:Esophageal squamous cell carcinoma;GC:Gastric cancer;MDR:Multidrug resistance;MSCs:Mesenchymal stem cells;TGF-β:Transforming growth factor-β;TβRI:Type 1 TGF-β receptor;TβRII:Type 2 TGF-β receptor.
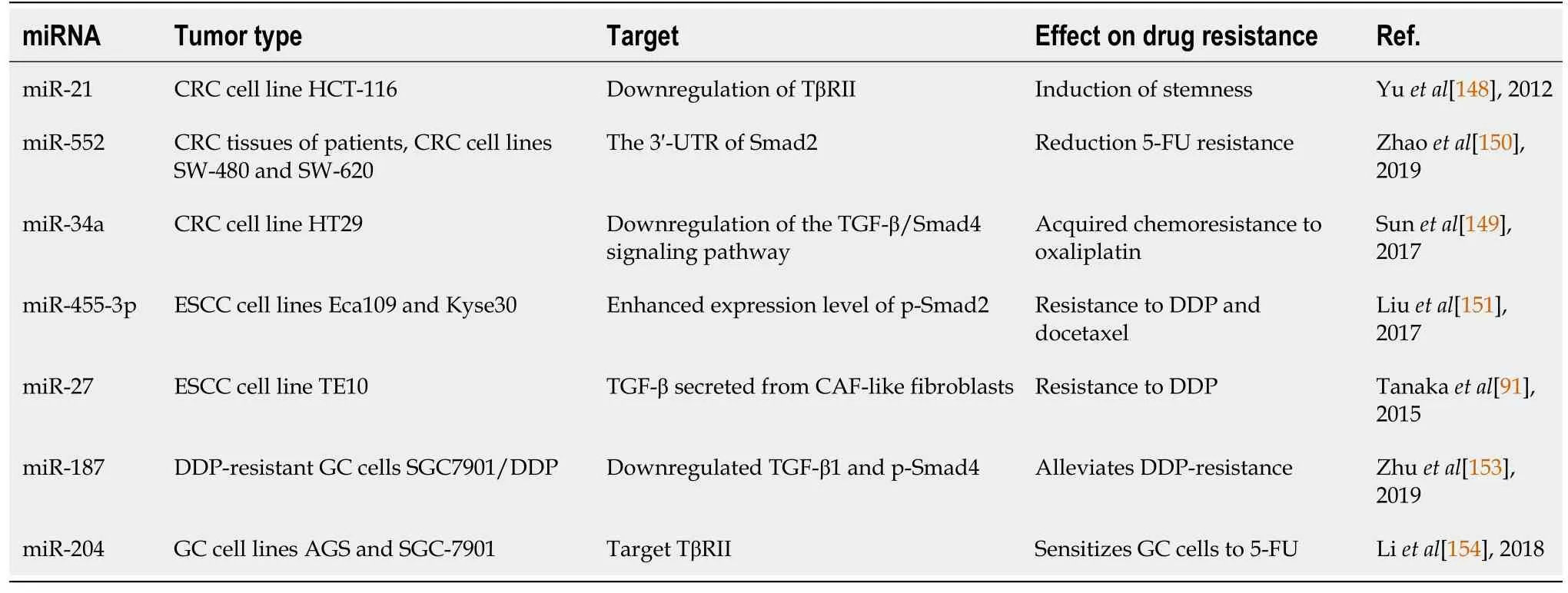
Table 2 Studies evaluating the relationship between miRNAs and drug resistance related to the transforming growth factor-β signaling pathway in gastrointestinal cancer
Conventional chemotherapy
Fluorouracil:5-FU belongs to the antimetabolite family[70]and is a commonly used chemotherapeutic regimen for CRC and GC.5-FU,a pyrimidine analog and an inhibitor of thymidylate synthase,is incorporated into RNA or DNA in the place of uracil or thymine and leads to the prevention of DNA replication and cell death[71].Unfortunately,the treatment effectiveness of 5-FU is reduced,and its clinicalapplication is limited by the emergence of drug resistance.The response rate to 5-FU is limited to 10%–15% in CRC.Various strategies have been used to improve the efficacy of 5-FU,resulting in the extension of the median survival to 30 mo[72].
A study of colorectal tumor biopsies shows that CRC patients with normal Smad4 diploidy experienced a threefold higher benefit from postoperative 5-FU-based adjuvant chemotherapy than those with Smad4 deficiency[73].Another study of the same collection of tumor specimens reveals that serine-threonine receptor-associated protein,a TGF-β-signaling inhibitor that acts at the receptor level,was a predictor of unfavorable responses to 5-FU-based adjuvant chemotherapy[74].Similarly,CRC patients treated with surgery and 5-FU-based adjuvant therapy and followed for over 6 years to evaluate the prognostic value of Smad4 expression,demonstrate that patients with a low level of Smad4 expression had shortened DFS and OS compared with those with a high level of Smad4 expression[75].In HCT116 colon cancer cells,Smad4 deficiency is found to be responsible for 5-FU resistance[76].Moreover,in an azoxymethane rat model of colon cancer,vitamin D3 supplementation promotes the efficacy of 5-FU through multiple mechanisms including increased expression of TGFβ1,TβRII,and Smad4[77].In brief,the results indicate that the effectiveness of adjuvant 5-FU-based chemotherapy might depend on TGF-β signaling in CRC.
As the TGF-β signaling pathway appears to have both suppressive and promoting effects in cancer,other studies have suggested that activation of the TGF-β signaling pathway might induce resistance to 5-FU in GI cancers.Immunohistochemical staining in patients with stage II-III advanced rectal cancer showed that p-Smad3 overexpression was associated with poor preoperative responses to fluoropyrimidine-based chemoradiotherapy.Therefore,p-Smad3 could be a potential predictor of a poor response to radiochemotherapy[78].Moreover,pre-CCRT serum TGF-β1 levels were found to be negatively correlated with DFS in patients with ESCC receiving concurrent neoadjuvant chemoradiotherapy with taxane-based/5-FU-based chemotherapy followed by esophagectomy[79].In CRC cells,TGF-β1 treatment was found to increase apoptotic resistance in cells exposed to therapeutics including 5-FU and etoposide[80].TGF-β inhibition was found to sensitize HCT116 cells to 5-FU treatment and suppress cell migration[81].Likewise,TβRI inhibition reduced proliferation and increased cell death in chemoresistant cancer cells[82].Furthermore,Moonet al[83]found that Smad3/4 acted as a drug sensitivity regulator in TGF-β-mediated chemoresistant CRC cells,and knockdown of Smad3/4 significantly decreased tumor propagation and migration in the presence of 5-FU[83].In GC,hyaluronan-mediated motility receptor is a key regulator of chemoresistance,and its upregulation was found to promote EMT and CSC properties by activating the TGF-β/Smad2 signaling pathway,ultimately leading to 5-FU resistance[84].
Platinum compounds:Platinum compounds are used as single agents or in combinatorial regimens for the treatment of GI cancers.The molecular mechanism of platinum compound-induced apoptosis involves the inhibition of DNA synthesis and repair,resulting in cell cycle arrest.This effect is mediated by the activation of various signal transduction pathways[85].OXA is an important platinum-based option for the treatment of CRC[86].In two multicenter trials in which single-agent OXA was administered as first-line treatment of advanced CRC,response rates were 12% and 24%,progression-free survival was 4 mo,and median survival was 14.5 mo and 13 mo,respectively[87].
In CRC cells,TGF-β1 contributes to OXA resistance primarily through EMT,which leads to antiapoptotic effects and the attenuation of DNA damage[88].Furthermore,both siRNA-mediated knockdown of Smad2/3 and treatment with the potent TGF-β inhibitor SB43154225 suppress migration and invasion and increase therapeutic sensitivity to OXA in HCT116 and DLD1 CRC cell lines[89].Curcumin,a naturally occurring polyphenolic substance extracted from the Curcaceae plantCurcuma longa,sensitizes CRC to OXA treatment by inhibiting the TGF-β/Smad2/3 pathway in the OXA-resistant cell line HCT116/OXA and in anin vivoanimal model of CRC[90].In EC,TGF-β secreted from CAF-like fibroblasts induces chemoresistance to cisplatin,which is reversed after administration of TGF-β neutralizing antibodies[91].
Taxoid compounds:Paclitaxel(PTX)is an antineoplastic agent derived from the bark of the Pacific yewTaxus brevifolia[92].Docetaxel is a semi-synthetic taxane that primarily acts to promote microtubule assembly and prevents the depolymerization of assembled microtubules[93].Both PTX and docetaxel exert potent antitumor effects by stabilizing microtubules,resulting in cell cycle arrest and apoptosis[94].The results of a multicenter trial in patients with advanced or recurrent GC showed that the response rate to PTX as a second-line monotherapy was 17.5%[95].The median duration of response to PTX monotherapy was 2.8 mo in patients with advanced gastric or gastroesophageal junction adenocarcinoma,and the patients eventually developed resistance to PTX[96].The results of a phase II study in previously-untreated GC patients reported overall response rates to single-agent docetaxel in the range of 17% to 24%[97].
Peritoneal dissemination is the most common mode of metastasis in GC.Low-dose PTX can significantly inhibit Smad2 phosphorylation in human peritoneal mesothelial cells,leading to a decrease in stromal fibrosis[98].The results of a microarray analysis showed that C-X-C chemokine receptor type 4(CXCR4)was a novel marker for highly metastatic CSCs.Treatment with TGF-β enhanced the anticancer effect of docetaxelviathe induction of cell differentiation/asymmetric cell division within the CXCR4-positive gastric CSC population,even when the cells were in a dormant state[99].Bone morphogenetic protein 4(BMP-4),which is involved in TGF-β signaling,is upregulated in PTX-resistant human esophageal carcinoma EC109 cells and docetaxelresistant human GC MGC803 cells.p-Smad1/5,which is also involved in the TGF-β/Smad pathway,is also overexpressed in EC109/Taxol cells[100].
Doxorubicin:Doxorubicin(Dox),a chemotherapeutic agent extensively used to treat a wide range of cancers,exerts cytotoxic and DNA damaging effects through interference with nucleoside metabolism,but is less efficacious in GI cancers relative to other cancer types[101].The antineoplastic activity of Dox is attributed to its intercalation into the DNA helix and its ability to generate free radicals[102].In HCT116 colon cancer cells,long-term administration of low concentrations of Dox may promote resistance partlyviathe activation of TGF-β signaling.Moreover,knockdown of Smad4 significantly increases the sensitivity of HCT116 cells to Dox,in partviathe inhibition of multidrug-resistant plasma membrane glycoprotein expression and reversal of the EMT process[103].Therefore,the combination of Dox treatment and TGF-β downregulation might be a potential therapeutic strategy to overcome chemoresistance.
Adriamycin:Adriamycin(ADM)generates superoxide radicals that kill tumor cells by damaging DNA,directly intercalating into DNA,and preventing DNA replication.In human EC cells(T.T)and GC cells(MKN28 and MKN45),pretreatment with TGF-β1 results in increased sensitivity to ADM.In vivo,the combined administration of TGF-β 1 and ADM delayed tumor growth better than either treatment alone and further exhibited synergistic antitumor effects[104].
Targeted therapy
Knockdown of MED12 in the CRC cell lines SK-CO-1(KRASV12)and SW1417(BRAFV600E)resulted in the activation of MEK/ERK and induced resistance to the MEK inhibitor AZD6244(selumetinib).Moreover,TGF-β-induced resistance to AZD6244 and the BRAF inhibitor PLX4032(vemurafenib)have also been observed in CRC cells[67].However,another study demonstrated that vemurafenib downregulated the expression of TGF-β and p-Smad3 in HT29 CRC cells[105].Trastuzumab,a human epidermal growth factor receptor(HER)2-targeting antibody,is the only available targeted agent for first-line palliative systemic treatment of HER2-positive esophagogastric adenocarcinoma(EAC).EAC cells become resistant to trastuzumab and the HER2-HER3 signaling inhibitor pertuzumab by activating TGF-β signaling,which subsequently induces EMT.TGF-β receptor inhibitors were shown to increase the antitumor efficacy of trastuzumab and pertuzumab in EAC cells and EAC patientderived xenograft tumors[106].Sensitivity of the GC cell line NCI-N87 to trastuzumab was significantly decreased after treatment with TGF-β.Moreover,TGF-β was upregulated in trastuzumab-resistant NCI-N87/TR cells[107].Cetuximab and trastuzumab,humanized antibodies against the HER family,exert antitumor effects by directly inhibiting epidermal growth factor receptor(EGFR)tyrosine kinase activity,inhibiting cell cycle progression,and activating proapoptotic molecules[108].In addition,an anti-TGF-β2 neutralizing mAb enhances cetuximab-mediated and trastuzumab-mediated antibody-dependent cellular cytotoxicity(ADCC)in TE1 TGF-β-producing ESCC cells.The TGF-β signaling inhibitor SB-431542 was found to enhance trastuzumab-mediated ADCC of TE1 cells.Furthermore,the exogenous addition of TGF-β2 significantly decreased cetuximab-mediated ADCC in non-TGF-β2-producing TE5 cells,and TGF-β2 inhibited the activity of trastuzumab-mediated ADCC in TE1 cells[109,110].TGF-β expression is upregulated in three FGFR2-amplified SNU-16 GC cell lines that are resistant to AZD4547,BGJ398,and PD173074.However,parental SNU-16 cells treated with TGF-β1 did not undergo EMT,and inhibition of TβRI was not sufficient to reverse EMT in the resistant cells[111].Bromodomain and extraterminal domain protein inhibitors(BETis)are in clinical trials as a novel class of cancer therapeutics.Both TβRII knockdown and treatment with the small-molecule Tβ RI inhibitor LY2157299(galunisertib)were reported to increase the sensitivity of RKO colon carcinoma cells to BETis[112].
Immunotherapy
Treatment with the TGF-β inhibitors P144 and P17 may be able to enhance the efficacy of immunotherapies by increasing antitumor immune responses[113].Moreover,treatment with the TGF-β-neutralizing mAb 1D11 enhanced the abscopal effect of radiotherapy as well as overall treatment efficacy in subcutaneous large MC38 colorectal tumors in conjunction with anti-programmed cell death protein 1(PD-1)plus anti-CD137 mAb[114].In mice with progressive metastatic liver disease,enabling immune infiltration using TGF-β inhibitors render tumors susceptible to anti-PD-1/Programmed cell death ligand 1(PD-L1)checkpoint-based therapies[115].Immunotherapies directed against TGF-β signaling may have broad applications in treating patients with advanced CRC.
Traditional medicine
Traditional herbal medicine has an important role in reversing the resistance of CRC cells to 5-FU.Hedyotis diffusaWilld,a traditional Chinese herbal medicine in the family of Rubiaceae,may exert its antimetastatic activity by suppressing TGF-β/Smad4 signaling pathway-mediated EMT in 5-FU-resistant CRC cells[116].Similarly,the traditional Chinese medicine formula Pien Tze Huang can effectively overcome multidrug resistance and inhibit EMTviasuppression of the TGF-β pathway in the 5-FU-resistant CRC cell line HCT-8/5-FU[117].Moreover,(1S,2S,3E,7E,11E)-3,7,11,15-Cembratetraen-17,2-olide(LS-1),a marine cembrenolide diterpene fromLobophytumsp.,can restore TGF-β signaling pathway activity and induce apoptosis in fluorouracilresistant human colon cancer SNU-C5/5-FU cells[118].
Various other Chinese herbs have been reported to exert antitumor or synergistic antitumor effectsviaTGF-β signaling pathway-mediated mechanisms.Oxymatrine,an alkaloid extracted from the Chinese herbSophora flavescensAit,can exert antimetastatic and anti-invasive effects through the inhibition of Smad2 phosphorylation and the formation of Smad2/3/4 in colorectal carcinoma RKO cells[119].Garcinol,a natural compound extracted fromGambogic genera,can inhibit EC metastasis in vitro andin vivoby dose-dependent suppression of p-Smad2/3 expression in the nucleus[120].In addition,a glycoprotein from the green algaCapsosiphon fulvescenswas shown to suppress the proliferation and migration of AGS GC cells by downregulating integrin expressionviainhibition of the TGF-β1-activated FAK/PI3K/AKT pathways[121].However,combination therapy with 5-FU and thymoquinone,which is the main bioactive compound derived fromNigella sativa,enhanced antitumor effects in a preclinical rat model of colorectal tumorigenesis partly by upregulating the expression of TGF-β1,TβRII,and Smad4[122].
TGF-β SIGNALING AND EMT IN GI CANCER DRUG RESISTANCE
TGF-β secreted from tumor cells is involved in paracrine signaling cascades that promote EMT and activate CAFs.CAFs,in turn,secrete more TGF-β that further drives EMT.Extracellular TGF-β binds to its receptor,resulting in the expression of key EMT genes.Furthermore,TGF-β can promote non-Smad pathways to accelerate EMT progression[16].It has been reported that fibronectin,a marker of EMT progression,induced EMT through Smad3/4-mediated TGF-β signaling[123].Therefore,TGF-β is an important inducer of EMT.SW837 rectal cancer cells treated with a TβR inhibitor or transfected with TβRII siRNA exhibited downregulation of mesenchymal markers,such as N-cadherin and vimentin and EMT regulators,including Snail,Twist,Slug,and Zeb1[124].Ginsenoside Rb2,the bioactive component in ginseng,inhibited EMT in CRC cells by inhibiting the expression of Smad4 and p-Smad2/3[125].Similarly,eribulin significantly inhibited EMT by downregulating the TGF-β/Smad pathway in GC[126].The EMT phenotype has been observed in GC cell lines resistant to 5-FU and AZD4547 and CRC cell lines resistant to BGJ398,PD173074,and OXA[84,111,127].Anticancer drugs can activate the TGF-β signaling pathway and further induce EMT,which is closely associated with chemotherapy resistance and evasion of immune surveillance[10,128].Dox treatment of HCT116 colon cancer cells was found to increase TGF-β1 and p-Smad2/3 expression and induce an EMT phenotype,exemplified by a reduction in E-cadherin and the upregulation of vimentin and N-cadherin.The changes ultimately resulted in the acquisition of Dox resistance.Furthermore,silencing Smad4 by stable RNA interference reversed the EMT process and increased the sensitivity of HCT116 cells to Dox[103].In EAC cells,EMT has been identified as a chemoradiation resistance mechanism in which EMT is mediated by the autocrine production of TGF-β in response to chemoradiation.Neutralization of TGF-β ligands effectively counteracted chemoradiation-induced EMT by reversing the mesenchymal phenotype[129].EAC cells incubated with trastuzumab and pertuzumab can secrete ligands for the TGF-β receptor and induce EMT-related changes,including reduced expression of epithelial markers(CD24,CD29,and CDH1)and increased expression of mesenchymal markers(CXCR4,VIM,ZEB1,SNAI2,and CDH2),resulting in drug resistance.However,combining the drugs with a TGF-β receptor inhibitor caused the cells to regain an epithelial phenotype[106].
TGF-β SIGNALING AND CSC IN GI CANCER DRUG RESISTANCE
Emerging evidence indicates that CSCs are the main factor underlying therapeutic failure,and chemotherapeutic resistance.The TGF-β pathway has been identified as a major stem cell-associated signaling pathway.ESCC has been found to arise from CSCs.Zhaoet al[130]showed that the TGF-β signaling pathway contributed to the lymphoid enhancer-binding factor 1-mediated CSC-like phenotype in ESCC cells.In EC,the TGF-β1 inhibitor SB525334 significantly suppressed the migration and invasion of sphere-forming stem-like cells,which possess key traits of CSCs,including chemoresistance[131].EMT is a critical process for the generation and maintenance of CSCs and the invasive front of ESCC.Moreover,the EGFR inhibitors erlotinib and cetuximab can both markedly suppress CSCs enrichmentsviaTGF-β1-mediated EMT in ESCC[132].In mouse GC cells,activation of the TGF-β pathway downregulated the expression of Sca-1,which has been identified as a potential CSC enrichment marker.High expression of Sca-1 was related to increased resistance to cisplatin/fluorouracilbased chemotherapy[133].In addition,TGF-β enhanced the anticancer effect of docetaxel by inducing the differentiation of gastric CSCs[99].
TGF-β SIGNALING AND TUMOR MICROENVIRONMENT IN GI CANCER DRUG RESISTANCE
TGF-β is a pleiotropic cytokine with potent immunosuppressive effects.TGF-β downregulates CD8+and CD4+T cell activation and stimulates the differentiation of immune-suppressive regulatory T(Treg)cells[10,114].CRC cells secrete anti-inflammatory cytokines,including TGF-β,which can affect the dendritic cell(DC)phenotype and support tumor escape from immune surveillance[134].However,the TGF-β receptor inhibitor SB-431542 can induce potent phenotypic and functional maturation of DCs and trigger an antitumor immune response[135].In ESCC,TGF-β1 was shown to partially contribute to the downregulation of CD16 on natural killer(NK)cells,resulting in NK cell dysfunction[136].
TGF-β signaling pathway activation plays an important role in immune evasion and contributes to immune checkpoint therapy failure[137,138].Enabling immune infiltration by blocking TGF-β signaling renders tumors susceptible to anti-PD-1-PD-L1 checkpoint-based therapy[115].Moreover,the TGF-β neutralizing monoclonal antibody 1D11 markedly enhanced the abscopal effects and the overall treatment efficacy in conjunction with an anti-PD-1 plus anti-CD137 mAb combination in large MC38 colorectal tumors[114].In ESCC,myeloid-derived suppressor cell-derived TGFβ increased PD-1 expression on CD8+T cells,which led to resistance to PD-1/PD-L1 blockade in the tumor microenvironment.Dual PD-1/PD-L1 and TGF-β pathway blockades restored the function and antitumor ability of CD8+T cells[139].Furthermore,combined treatment with cyclophosphamide and interleukin(IL)-12-expressing adenovirus,which might be a valid immunotherapeutic strategy for advanced GI cancer,was shown to revert the Treg immunosuppressive phenotype by blocking the secretion of IL-10 and TGF-β,resulting in loss of their DC inhibitory activity[140].
CAFs are the most abundant cell type in the tumor microenvironment.One of the main sources of CAFs is endothelial cells undergoing EMT,which is mainly promoted by TGF-β[141].CAFs can confer TGF-β1-mediated ESCC cell resistance to several chemotherapeutic drugs,including cisplatin,taxol,irinotecan,5-FU,carboplatin,docetaxel,pharmorubicin,and vincristine.Inhibition of CAF-secreted TGF-β1 signalingviatreatment with the TβRI inhibitor LY2157299 significantly enhanced chemosensitivity[142].Moreover,TGF-β secreted by miR-27-induced CAFs induced chemoresistance to cisplatin in EC[91].In CRC,Snail-expressing 3T3 fibroblasts exhibit CAF properties that support 5-FU and PTX chemoresistanceviaTGF-β/NF-κBmediated CCL1 secretion[143].Tanget al[144]found that,in CRC,hypoxia-inducible factor 1α(HIF-1α)and CAF-secreted TGF-β2 synergistically induced the expression ofGLI2,which promoted chemoresistance.
Mesenchymal stem cells(MSCs),an important part of the tumor environment,contribute to the development of drug resistance[145].In GC cells,TGF-β1 secretion by MSCs activated Smad2/3 and induced expression of the lncRNA MACC1-AS1 that promoted FA oxidation-dependent stemness and chemoresistance to 5-FU and OXA[146].
TGF-β SIGNALING AND MIRNA IN GI CANCER DRUG RESISTANCE
Emerging evidence indicates that some miRNAs can regulate the resistance of GI cancers to a variety of chemotherapeutic drugs through the TGF-β signaling pathway,as summarized in Table 2.In HT-29 colon cancer cells,overexpression of miR-146a was found to be associated with various processes in the cancer microenvironment,including enhancement of 5-FU and irinotecan resistance and promotion of TGF-β secretion[147].MiR-21 was shown to increase both stemness and the overall proportion of CSCs in colon cancer cells by downregulating TβRII,a direct target of miR-21,and by activating the Wnt/β-catenin pathway[148].MiR-34a was found to mediate OXA resistance in CRC cells by inhibiting macroautophagyviaregulation of the TGF-β/Smad4 pathway[149].However,the expression levels of miR-552 were negatively correlated with resistance to 5-FU-based chemotherapy in CRC cells.Mechanically,miR-552 directly targeted the 3'-UTR of Smad2,and stable knockdown of Smad2 reversed miR-552 deficiency-induced 5-FU resistance[150].Overexpression of miR-455–3p conferred resistance to cisplatin and docetaxel in ESCC cells,whereas miR-455–3p antagonism reversed chemoresistance and reduced the number of CD90+and CD271+tumor-initiating cellsviathe suppression of multiple stemness-associated pathways,including TGF-β signaling[151].Moreover,miR-27 has shown to play a role in cisplatin resistance in EC through the transformation of normal fibroblasts into CAFs and the induction of TGF-β secretion from the CAFs[91].In GC,overexpression of miR-577 contributed to TGF-β-mediated EMT and stemness by forming a positive feedback loop,resulting in chemoresistance to OXA[152].However,overexpression of miR-187 in GC cells alleviated cisplatin resistance by inhibiting the TGF-β/Smad signaling pathway[153].Furthermore,overexpression of miR-204 was found to sensitize 5-FU-resistant GC cells through the suppression of TβRII-mediated EMT[154].
CONCLUSION
Drug resistance,which leads to unfavorable clinical outcomes and treatment failure,remains a considerable challenge in the treatment of GI cancers.The TGF-β signaling pathway plays an important role in the regulation of the drug responses to conventional chemotherapy,targeted therapy,immunotherapy,and traditional medicine.Furthermore,TGF-β-mediated drug resistance in GI cancers is closely associated with several processes,including EMT,CSC development,alteration of the tumor microenvironment,and miRNA biogenesis(Figure 1).
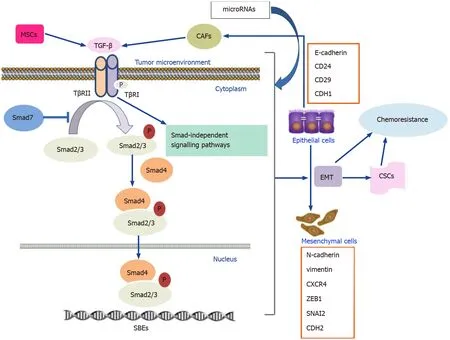
Figure 1 Mechanisms of transforming growth factor-β signaling and involvement in gastrointestinal cancer chemoresistance.CAFs:Cancer-associated fibroblasts;CSCs:Cancer stem cells;EMT:Epithelial-mesenchymal transition;MSCs:Mesenchymal stem cells;TβRI:TGF-β Type 1 receptor;Tβ RII:TGF-β Type 2 receptor.
Despite improvements in treatment strategies,EC,GC,and metastatic CRC have a poor prognosis,with 5-year OS rates of 15%–25%,29.3%,and 14%,respectively[2,155,156].The key obstacle to therapeutic success is the development of drug resistance,highlighting the urgency driving the development of alternative treatments for GI cancers.Many reports indicate the benefits of combining antitumor agents with agents that suppress TGF-β signaling.However,the findings require further verification by additional clinical studies.The use of some small-molecule inhibitors of TGF-β signaling is currently being investigated in both preclinical and clinical trials[60,157].As TGF-β possesses paradoxical activities,the identification of potential biological markers related to the response to TGF-β inhibitors would have important clinical implications and would help select patients most likely to benefit from their use.
 World Journal of Gastrointestinal Oncology2021年11期
World Journal of Gastrointestinal Oncology2021年11期
- World Journal of Gastrointestinal Oncology的其它文章
- Hepatocellular carcinoma biomarkers,an imminent need
- Anatomical vs nonanatomical liver resection for solitary hepatocellular carcinoma:A systematic review and meta-analysis
- Atezolizumab plus bevacizumab versus sorafenib or atezolizumab alone for unresectable hepatocellular carcinoma:A systematic review
- Cell-free DNA liquid biopsy for early detection of gastrointestinal cancers:A systematic review
- Colorectal cancer in Arab world:A systematic review
- Induction chemotherapy with albumin-bound paclitaxel plus lobaplatin followed by concurrent radiochemotherapy for locally advanced esophageal cancer