
Role of mammalian target of rapamycin complex 2 in primary and secondary liver cancer
2021-11-20 06:33:08KatharinaJoechleJessicaGuenzleClausHellerbrandPavelStrnadThorstenCramerUlfPeterNeumannSvenArkeLang
Katharina Joechle,Jessica Guenzle,Claus Hellerbrand,Pavel Strnad,Thorsten Cramer,Ulf Peter Neumann,Sven Arke Lang
Katharina Joechle,Thorsten Cramer,Ulf Peter Neumann,Sven Arke Lang,Department of General,Visceral and Transplantation Surgery,University Hospital Rheinisch-Westfälisch Technische Hochschule Aachen,Aachen 52074,Germany
Jessica Guenzle,Department of General and Visceral Surgery,Medical Center-University of Freiburg,Faculty of Medicine,University of Freiburg,Freiburg 79106,Germany
Claus Hellerbrand,Institute of Biochemistry,Friedrich-Alexander University Erlangen-Nürnberg,Erlangen 91054,Germany
Pavel Strnad,Department of Internal Medicine III,University Hospital Rheinisch-Westfälisch Technische Hochschule Aachen,Aachen 52074,Germany
Abstract The mammalian target of rapamycin(mTOR)acts in two structurally and functionally distinct protein complexes,mTOR complex 1(mTORC1)and mTOR complex 2(mTORC2).Upon deregulation,activated mTOR signaling is associated with multiple processes involved in tumor growth and metastasis.Compared with mTORC1,much less is known about mTORC2 in cancer,mainly because of the unavailability of a selective inhibitor.However,existing data suggest that mTORC2 with its two distinct subunits Rictor and mSin1 might play a more important role than assumed so far.It is one of the key effectors of the PI3K/AKT/mTOR pathway and stimulates cell growth,cell survival,metabolism,and cytoskeletal organization.It is not only implicated in tumor progression,metastasis,and the tumor microenvironment but also in resistance to therapy.Rictor,the central subunit of mTORC2,was found to be upregulated in different kinds of cancers and is associated with advanced tumor stages and a bad prognosis.Moreover,AKT,the main downstream regulator of mTORC2/Rictor,is one of the most highly activated proteins in cancer.Primary and secondary liver cancer are major problems for current cancer therapy due to the lack of specific medical treatment,emphasizing the need for further therapeutic options.This review,therefore,summarizes the role of mTORC2/Rictor in cancer,with special focus on primary liver cancer but also on liver metastases.
Key Words:Mammalian target of rapamycin;Mammalian target of rapamycin complex 2;Rictor;Liver cancer;Liver metastases;Hepatocellular carcinoma;Cholangiocellular carcinoma
INTRODUCTION
The mammalian target of rapamycin(mTOR)is an atypical serine/threonine kinase that controls cell survival,proliferation,and metabolism through phosphorylation of its downstream targets.It acts in two structurally and functionally distinct protein complexes,mTOR complex 1(mTORC1)and mTOR complex 2(mTORC2),and can be activated by several stimulating factors as hypoxia,insulin,growth factors,or dysregulation of PI3K/Akt signaling[1,2].However,upon deregulation,activated mTOR signaling is implicated in the hallmarks of cancer and associated with increased cell survival,uncontrolled cell proliferation,metabolic reprogramming,and aberrant angiogenesis[3],which makes it a promising target in anticancer therapy.
The incidence of primary liver cancer,such as hepatocellular carcinoma(HCC)and intrahepatic cholangiocellular carcinoma(iCCC),is increasing worldwide.Systemic therapy options are limited for both cancer entities.Surgery offers the only chance for cure,although only a minority of patients with HCC or iCCC are eligible for resection.Moreover,the liver is one of the most frequent sites of metastases development.Indeed,in most cancer entities liver metastasis is associated with a dramatic decline of patient’s prognosis,further emphasizing the fundamental impact of this site.A deeper understanding of processes involved in either primary or secondary liver cancer is therefore urgently needed[4-6].
Due to the availability of(more or less)selective mTORC1 targeting agents such as rapamycin,the role of mTORC1 in cancer has been extensively studied for decades.In contrast,mTORC2 was less intensively analyzed,mainly because of the lack of selective pharmacologic inhibitors.However,the last decade has brought up several studies suggesting a role for mTORC2 in cancer.For instance,Rictor(rapamycin insensitive companion of TOR),the central subunit of mTORC2,was found to be upregulated in different kinds of cancer and is associated with impaired prognosis[7-10].Evidence from primary and secondary liver cancer is summarized in Table 1.In addition,involvement of mTORC2/Rictor in a plethora of processes implicated in tumor growth and also metastasis have been reported[11].Therefore,the present review summarizes the current knowledge regarding the role of mTORC2 in cancer with focus on primary and secondary liver cancer.
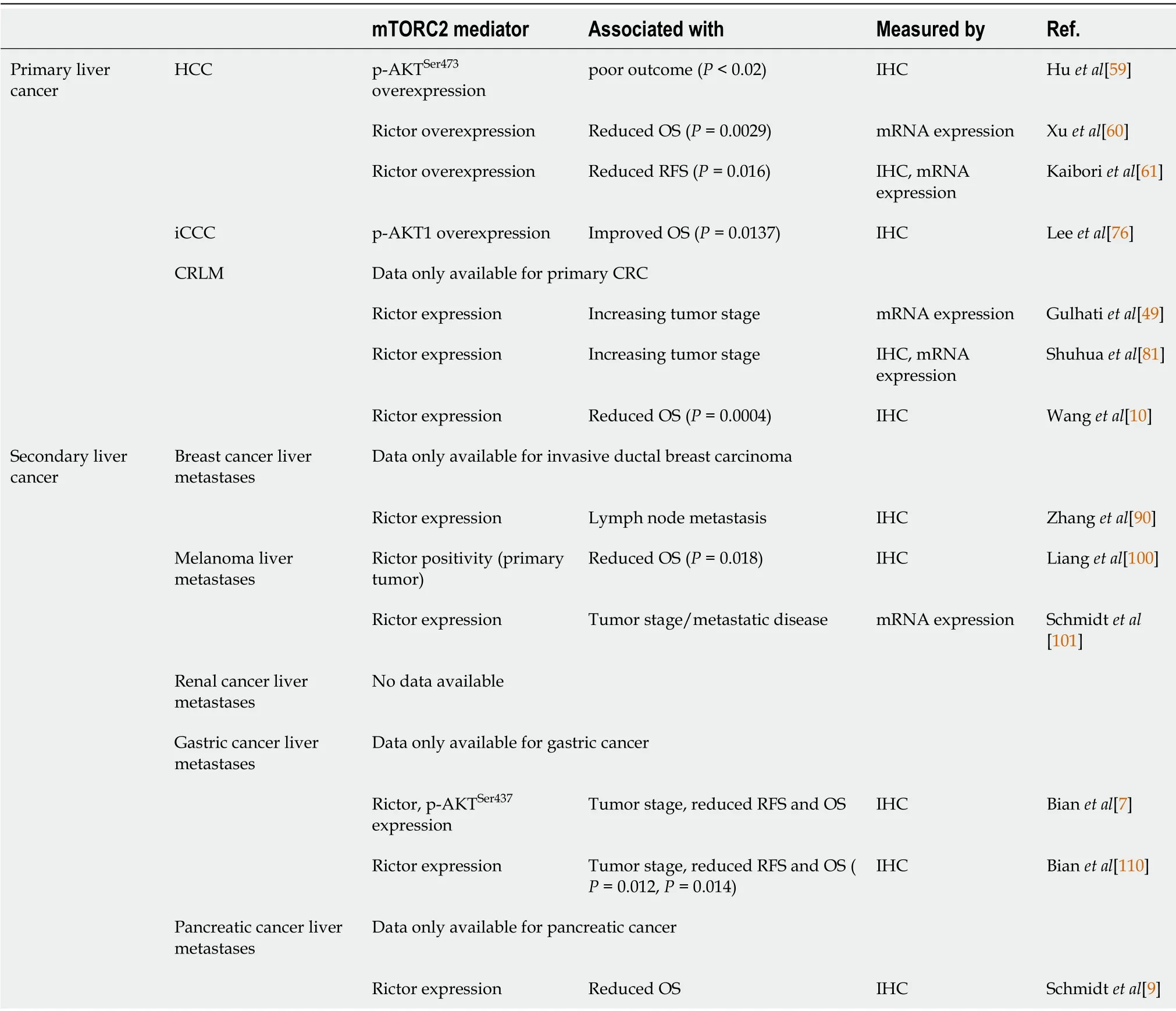
Table 1 Overexpression of mammalian target of rapamycin complex 2 and its mediators determine clinical outcome
MTOR COMPLEXES AND SIGNALING
Although mTOR acts through the different complexes mTORC1 and mTORC2,both have several subunits in common:the catalytic kinase mTOR,the scaffolding proteinmLST8,the regulatory subunit DEPTOR,and the stabilizing complex Tti1/Tel2.The distinct subunits of mTORC1 are Raptor and PRAS40.While Raptor is important for mTORC1 substrate specificity,stability,and regulation,PRAS40 acts as negative regulator of mTORC1.Activation of mTORC1 depends on nutrient(e.g.,amino acids)and growth factor(e.g.,insulin)signaling through the PI3K/AKT and Ras-MAPK cascades.Phosphorylated AKT(viathe PI3K/AKT pathway)or ERK and RSK(viathe Ras-MAPK cascade)inhibit the TSC1/2 complex,which in turn triggers RHEBmediated activation of mTORC1 leading to phosphorylation of its substrates 4EBP1 and S6K1.The downstream effectors act as key regulators of cap-dependent and capindependent mTORC1 translation[3]and regulate translation and transcription of different target genes(e.g.,HIF1α,etc.)thereby being implicated in cell growth,proliferation,and metabolism[12].
mTORC2 consists of its specific subunits Rictor and mSin1.Similar to Raptor,Rictor controls mTORC2 stability,subcellular localization,and substrate identification.It is essential for mTORC2 function,as silencing Rictor leads to significant inhibition of AKT,the key substrate of mTORC2.mSin1 negatively regulates mTORC2 until PI3Kmediated growth factor signaling locates mSin1/mTORC2 to the plasma membrane and relieves its inhibition.In turn,PI3K-generated PIP3 activates mTORC2.Activated mTORC2/Rictor leads to phosphorylation of AKT at the Ser473 residue.AKT can also be phosphorylated at the Thr308 residue by PDK1,which is in turn attracted by PIP3.Importantly,AKT is one of the most frequently activated proteins in cancer,and its full activity is only achieved if both sites are phosphorylated[13,14].On a functional level,AKT is involved in many processes.Association with cell migration,invasion,increased tumor growth,and cell survival while inhibiting apoptosis and promoting proliferative processes including glucose uptake and glycolysis have been described[15,16].Additional substrates of mTORC2 are the AGC kinases like the PKC(e.g.,PKC α,PKCδ,PKCε)family,which control tumorigenesis,cell migration,and cytoskeletal remodeling,and SGK isoforms that are implicated in cell survival and resistance to chemotherapy.
Along with the essential role of Rictor for mTORC2 functioning,it also acts independently of mTORC2.Thr1135,one of the 37 phosphorylation sites of Rictor,was shown to be stimulated directly by growth factor signaling and to be sensitive to rapamycin,as it is targeted by S6K1,one of the downstream effectors of mTORC1[17,18].This mechanism is assumed to represent a regulatory link between mTORC1 and mTORC2 signaling and to be part of a reciprocally influenced feedback loop mechanism[19,20].Moreover,Rictor was also described to be associated with complexes exhibiting oncogenic and tumor suppressor properties.For example,Rictor forms a complex with ILK,which is crucial for TGFß1 mediated epithelial-to-mesenchymal transition(EMT)and cancer cell survival[16,21].In contrast,the combination of Rictor with PCD4 acts in an anti-oncogenic manner,as renal cancer cells showed reduced metastatic ability[22].
IMPLICATIONS OF MTORC1 IN CANCER
Besides being implicated in many physiological processes such as glucose and lipid homeostasis,adipogenesis,maintaining muscle mass and function,brain and immune function,deregulation of mTORC1 signaling is not only associated with diseases such as diabetes,neurodegeneration,and cancer.As described above,mTORC1 is activated by the oncogenic pathways PI3K/Akt and Ras-MAPK cascade.However,the oncogenic pathways are frequently mutated,resulting in hyperactivation of mTORC1,which is found in many human cancers[1].Hyperactivated mTORC1 and its association with tumorigenesis are also seen in tuberous sclerosis,a familial cancer syndrome defined by the loss of the TSC1/2 complex,a negative regulator of mTORC1.Downstream of mTORC1,its role in carcinogenesis is linked to metabolic reprogramming in cancer cells.One example is the Warburg effect;aerobic glycolysis is controlled by mTORC1viaincreased translation of HIF1α that in turn regulates the expression of glycolytic enzymes[23].mTORC1 is also associated with upregulation of genes involved in lipogenesis by activation of the transcription factor SREBP1 through phosphorylation of Lipin1 and S6K1[24,25].The latter was shown to be a major mechanism to promote growth and proliferation in breast cancer cells[26].mTORC1-mediated phosphorylation of S6K1 is not only involved in lipogenesis but also in purine and pyrimidine synthesis leading to a rapid DNA duplication in cancer cells[27,28].Aside from its control in cancer cell metabolism,mTORC1 is also involved in the regulation of autophagy and macropinocytosis.
The implications of mTORC1 in cancer are widely studied because of the availability of rapamycin as a selective mTORC1 inhibitor.However,rapamycin analogs(rapalogs)have only shown limited efficacy in cancer therapy.While inhibition of mTORC1 by rapamycin blocks phosphorylation of S6K1,phosphorylation of 4EBP1 is not fully blocked[29].Therefore,4EBP1-regulated translation of proteins involved in tumorigenesis is not inhibited.Another reason for the limited efficacy of rapamycin and rapalogs is explained by compensatory upregulation of AKT through phosphorylation of its Thr308 and Ser473 residue[30,31]as inactivated S6K1 no longer prevents suppression of insulin-PI3K signaling[32,33].Moreover,treatment with rapalogs may increase micropinocytosis and autophagy leading to enhanced cell proliferation and survival[34,35].
IMPLICATIONS OF MTORC2 IN CANCER
Compared with mTORC1,much less is known about the role of mTORC2 in cancer,although existing data suggest mTORC2 to be of importance.Particularly,it is one of the key effectors of the PI3K/AKT/mTOR pathway and stimulates cell growth,cell survival,metabolism,and cytoskeletal organization.
Tumorigenesis
As described above,AKT is the key downstream target of mTORC2 signaling and one of the most commonly activated proteins in cancer[36,37]with its isoforms AKT1 and AKT2 being the main effectors in tumorigenesis[38].With more than 200 AKT substrates,the different isoforms seem to have unique roles in tumorigenesis.While AKT1 increases tumor development and reduces tumor invasion,the expression of AKT2 has the opposite effect[39].Frequently,hyperactivated AKT is found in different kinds of cancers.For example,hyperactivation of AKT caused by a somatic mutation was found to induce B-cell lymphoma,in contrast to wild-type AKT[40].The somatic mutation was also found in breast,colorectal,and ovarian cancer.In addition to somatic mutations,hyperactivation of AKT may also occur because of activating upstream mutations in PI3K or deletions in PTEN,a tumor suppressor[41].AKT has several substrates in common with SGK family members,another downstream target of mTORC2.Hence,SGK is similarly implicated in cell growth and proliferation[42].Its isoform SGK3 is dependent on PDK1 signaling to induce tumor growth and adopted the role of AKT in tumorigenesis in PI3K mutated cancer cells[43].AKT and SGK,which are different isoforms of PKC,are also involved in tumorigenesis.PKC-ε,PKC-λ/ι,and PKC-β are known oncogenes with PKC-λ/ι and PKC-β for example being involved in colon carcinogenesis[44,45].Mechanisms by which mTORC2 is involved in tumorigenesis are shown in Figure 1.
Metastasis
Cell migration and invasion are the two key components of metastasis that are affected by mTORC2 through different pathways.On the one hand,phosphorylation of AKT leads to activation of Rac1 through activation of Tiam1[46].On the other hand,Rac1 is also upregulated by the suppression of its inhibitor RhoGD12,not only through AKT but also independent of AKT through PKCα activation[46,47](Figure 2).Rac1 and RhoA are small GTPases known to have crucial roles in actin cytoskeletal rearrangement and cell migration,mainly by stimulating lamellipodia formation[48].Upon mTORC2 knockdown,expression of Rac1 and RhoA is decreased,leading to a reduction of colorectal metastasis[49].AKT1 is thus the AKT isoform implicated in metastasis.Silencing only AKT1 and not AKT2 reduces migration and invasion[50].The gain of invasive behavior is explained by the EMT,and was reversible upon mTORC2 and mTORC1 inhibition,which was followed by an increase in cell-cell contacts and E-cadherin;vimentin,SMA,fibronectin,and MMP9 decreased[49].
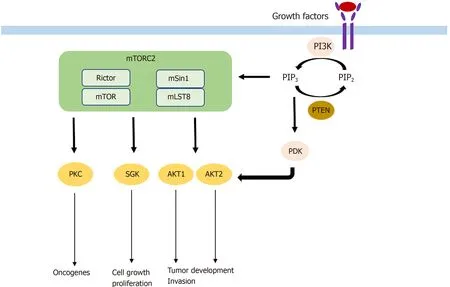
Figure 1 Mechanism by which mammalian target of rapamycin complex 2 participates in tumorigenesis.mTOR:Mammalian target of rapamycin;mTORC2:mTOR complex 2.
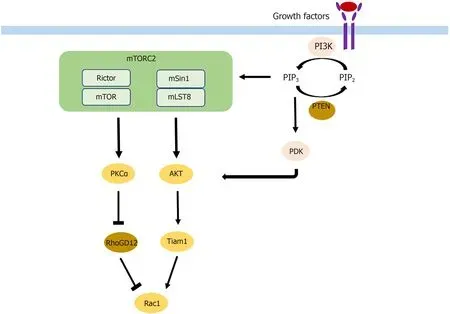
Figure 2 Mechanism by which mammalian target of rapamycin complex 2 participates in metastasis.mTOR:Mammalian target of rapamycin;mTORC2:mTOR complex 2.
Metabolic reprogramming
Metabolic reprogramming is a hallmark of cancer,and it allows tumor cells to receive and maintain their energy supply for rapid tumor growth[51].mTORC2 was shown to control c-Myc,a regulator of the Warburg effect,by phosphorylation of class IIa HDAC and acetylation of FoxO in both an AKT dependent and independent manner,thereby increasing glycolysis[52].Increases of glucose and acetate cause acetylation of Rictor,which in turn maintains mTORC2 signaling[53].Besides glycolysis,mTORC2 also controls cystine uptake and glutathione metabolism.Phosphorylation of SLC7A11 thereby allows tumor cells to focus mainly on survival rather than proliferation if the extracellular environment changes[54].In addition to energy supply,metabolic reprogramming can also be involved in drug resistance,as mTORC2 was shown to act as a central link between glucose metabolism and resistance to EGFR tyrosine kinase inhibitors[55].
Drug resistance
As mentioned above,metabolic reprogramming is one of the mechanisms of mTORC2-mediated drug resistance in cancer cells.Glucose metabolism has been linked not only to resistance to EGFR tyrosine kinase inhibitors[55]but also has caused Rictor acetylation that can be achieved by either glucose or acetate.Rictor acetylation induces auto-activation of mTORC2 signaling despite the absence of upstream growth factor signaling leading to resistance to EGFR-,PI3K- and AKT-targeted therapies[56].Furthermore,Rictor utilizes inhibition of apoptosis by activation of NF-κB as another mechanism to develop resistance to chemotherapy[57].Interestingly,the process could be overcome only by Rictor and not by AKT inhibition,suggesting that NF-κB is an AKT-independent mTORC2 downstream effector.In contrast,the positive feedback mechanism between amplified Rictor,known to occur in many cancers,and AKT leads to constant AKT activation,inducing not only tumor progression but also drug resistance[11].
MTORC2 IN PRIMARY LIVER CANCER
Hepatocellular carcinoma
HCC is the most common primary liver cancer and one of the main cancer-related deaths worldwide,with increasing incidence in recent years[58].Systemic treatment options,with the multikinase inhibitors sorafenib and lenvatinib as the only approved drugs,are very limited in case of unresectability or unavailability to local treatment options.Activation of mTORC2 as determined by immunohistochemistry of phospho-AKT was detectable in 60% of HCCs[59].Chromosomal gain of Rictor was described in 25% of HCCs,and its high expression was associated with a poor prognosis in HCC patients[60].Similarly,Kaiboriet al[61]found high expression of Rictor mRNA and protein and association with Rictor/Raptor ≥ 0,3 was a prognostic factor indicating poor recurrence-free survival.Rictor knockdown was shown to inhibit HCC cell growthin vitro[62],and AKT overexpressionin vivoled to increased HCC development[63].In the liver,there are two AKT isoforms,AKT1 and AKT2.Only AKT1 is phosphorylated and is thus activated by mTORC2 in c-Myc-induced HCC.AKT1 was the main driver of HCC formation,as AKT1 inhibition completely abolished c-Mycinduced tumor development[60].Silencing of Rictor also led to inhibition of c-Mycinduced HCC formation[60].In contrast,inhibition of AKT2 significantly reduced loss of PTEN-induced HCC formation[64].Moreover,it was found that loss of Rictor completely inhibited sgPTEN/c-Met HCC formation,leading to the assumption that mTORC2 regulates different AKT isoforms in HCC tumor development[65].In contrast,the role of the other mTORC2 substrates,PKC and SGK in HCC,remains poorly characterized.However,mTORC2 also impacts HCC tumorigenesis through its role in metabolic reprogramming.Fatty acid and lipid synthesis are triggered by mTORC2 thereby leading to hepatic steatosis and tumor development[66].The causal context was proven,as HCC development was completely abolished upon inhibition of fatty acid or sphingolipid synthesis[66].Not only mTORC2-associated lipid synthesis but also gluconeogenesis impacts HCC cell survival.Khanet al[67]showed that blocking mTORC2 led to increased gluconeogenesis and decreased HCC cell proliferation and survival.In addition to the important role of mTORC2 in hepatocarcinogenesis,it also seems to be involved in HCC metastasis and drug resistance.CHKA is an enzyme known to be associated with HCC metastasis and EGFRresistance.Inhibition of Rictor completely abolished CHKA-enhanced cell migration and invasion[8].In line with that,pharmacologic mTORC1/mTORC2 inhibition reduced tumor cell metastasis,while there was no effect shown if the mTORC1 inhibitor rapamycin was used[8].Moreover,following Rictor knockdown,CHKAmediated resistance of HCC cells to EGFR-inhibitors decreased,and sensitivity to the drugs increased[8].Similarly,the dual mTORC1/mTORC2 inhibitor OSI-027 reversed high MDR1 expression in HCC induced by doxorubicin and therefore increased chemosensitivity of doxorubicin.Combining both drugs led to inhibition of tumor growthin vitroandin vivo[68].In that context,the investigators concluded that mTORC2 was the component responsible for the effect,as inhibition of mTORC1 alone resulted in a modest decrease of MDR1 expression.
While these data show an important role of mTORC2/Rictor in the tumorigenesis and tumor progression of HCC,it is also involved in pre-tumor conditions.For example,Reyes-Gordilloet al[69]showed that the AKT isoforms were activated in anin vivotwo-hit model of alcoholic liver disease,leading to an increase of mTORC2 and inflammatory,proliferative,and fibrogenic genes.In line with the results,blocking of AKT1 and AKT2 led to a decrease in progression of liver fibrosis.In addition,mTORC2 was involved in the progression of nonalcoholic fatty liver disease(NAFLD)by dysregulation of white adipose tissue.Thereby,de novolipogenesis,lipolysis,glycolysis,and increased glucose uptake by GLUT4 are the mechanisms by which mTORC2 regulates adiposity and NAFLD[70].Besides alcoholic and nonalcoholic liver disease,viral hepatitis is one of the main risk factors for the development of HCC.In that context,increased AKT activity was demonstrated for hepatitis B and C.In hepatitis B,activation of AKT by the hepatitis B virus protein HBx leads to a persistent,noncytopathic virus replication[71].In hepatitis C,its NS3/4A protease increases AKT activity by enhancing EGF-induced signal transduction[72].
Intahepatic cholangiocarcinoma
iCCC is a highly aggressive tumor entity with increasing incidence in recent years[73].As systemic treatment only has partial benefits in advanced stages of iCCC[74],surgical resection remains the only curative option.Only a few studies examining the role of mTORC2 in iCCC exist.mTORC2 was found to be activated in almost 70% of iCCCs as determined by immunohistochemistry of phospho-AKT[75].When examining the AKT isoforms,protein expression of phospho-AKT1 was shown in 34% of patients with iCCC and was associated with a favorable prognosis[76].This unexpected result might be dependent on the mechanism of AKT activation triggering different downstream targets or a potential distinct role of AKT1 in iCCC.However,after Rictor knockdown,growth of iCCC cellsin vitrowas impaired and activated AKT was shown to cooperate with YAP to induce iCCC in mice[75].Moreover,in liverspecific Rictor knockout mice,cholangiocarcinogenesis induced by AKT/YapS127A was completely abolished,while wild-type mice had a lethal tumor burden at the same time point[75].Therefore,Zhanget al[77]used the pan-mTOR inhibitor MLN0128 and noticed significantly increased apoptosis but only slight effects on proliferation in iCCCin vitroandin vivo.Significantly enhanced apoptosis and consequently impaired cell proliferation in iCCC was also found after siRNA-mediated Rictor knockdown and simultaneous treatment with sorafenibviaincrease of FoxO1.Wanget al[78]reported another mechanism of tumorigenesis,which supported the oncogenic potential of mTORC2 signaling in iCCC.Briefly,activated AKT in combination with downregulation of the tumor suppressor FXBW7,increased cholangiocarcinogenesis.Interestingly,silencing cMyc in AKT/Fbxw7ΔF mice completely impaired iCCC growth[78].Furthermore,the results of a study by Yanget al[79]examining the impact of FXBW7 on EMT and metastasis of iCCC and perihilar CCC(pCCC)is also interesting even though it did not directly connect FXBW7 to mTORC2.In that study,silencing of FXBW7 lead to promotion of EMT,stem-like property,and metastasis of iCCC and pCCC.
While mTORC2 seems to be also involved in the pre-tumoral conditions of HCC including(non)alcoholic liver disease and viral hepatitis,no data exist on the role of mTORC2 chronic cholangitis,primary or secondary biliary cirrhosis as risk factors for the development of iCCC.In summary,mTORC2/Rictor seems to play a role in the development and progression of HCC and iCCCviadifferent mechanisms(Figure 3).However,more research is necessary to determine its exact role and to define potential targets for antineoplastic therapy.
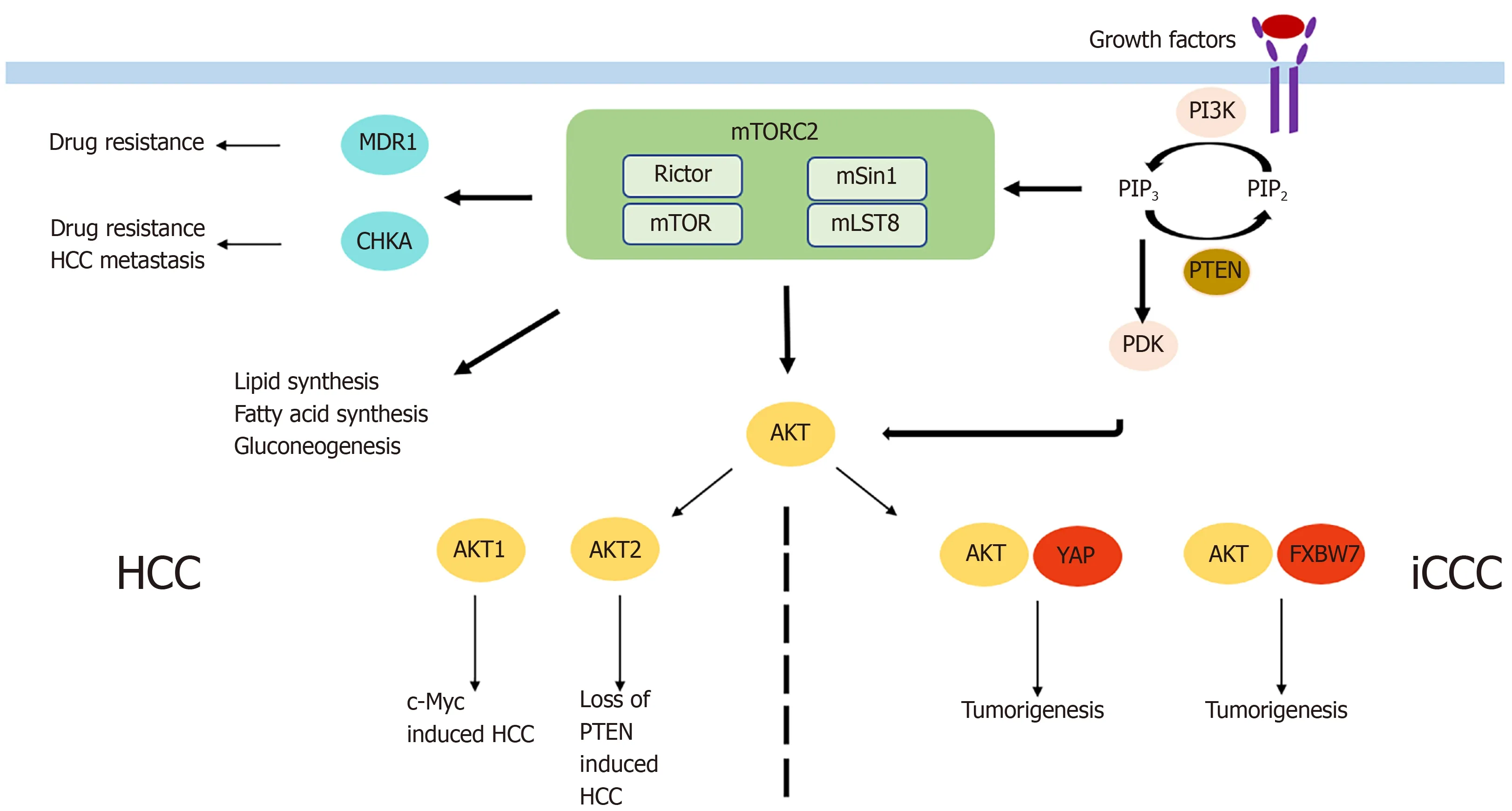
Figure 3 Mechanism by which mammalian target of rapamycin complex 2 is involved in tumorigenesis of primary liver cancer.HCC:Hepatocellular carcinoma;iCCC:Intrahepatic cholangiocellular carcinoma;mTOR:Mammalian target of rapamycin;mTORC2:mTOR complex 2.
MTORC2 IN SECONDARY LIVER CANCER
Colorectal cancer liver metastasis
Colorectal cancer(CRC)is the second leading cause of cancer-related deaths[80],with liver metastases being one of the most important predictors of poor long-term outcome.While it was shown that Rictor mRNA and protein are overexpressed in CRC[49]and expression is correlated with tumor progression,Dukes stage,lymph node metastasis,and impaired overall survival[10,81],there was no difference in Rictor expression between primary tumors and metastatic liver lesions[49].However,Rictor expression in primary tumors with metastatic liver lesions was significantly higher than it was in primary tumors without metastatic disease[49].Further,not only Rictor but also Raptor seems to be involved in colorectal liver metastases(CRLM),as knockdown of Rictor,as well as knockdown of Raptor,led to decreased migration and invasion of colorectal cancer cellsin vitro[49].In addition,in vivoknockdown of Raptor and Rictor in CRC cell lines impaired the formation of even micrometastases[49].A study by Gulhatiet al[49]did not focus on the development of liver metastases,but they showed that mTORC2viaRictor regulated actin cytoskeleton reorganization and cell migration through Rac1 and RhoA signaling.However,that is not the only mTORC2-associated mechanism involved in the formation of CRLM.TELO2,known to be essential for mTOR complex integrity,was found to be associated with colorectal tumorigenesis,migration,and invasion,as Rictor knockdown led to reduced TELO2-induced migratory and invasive behavior of colorectal cancer cells[82].Moreover,colorectal metastasis is not controlled onlyviaRictor but also by mSin1.Wanget al[83]showed that the tumor suppressor Pdcd4 inhibited Sin1 translation leading to reduced mTORC2 activation and inhibited invasion of CRC cells.While the studies support the important oncogenic and metastatic potential of mTORC2 in CRC,it is also involved in resistance to systemic chemotherapeutic agents[81].In particular,resistance of CRC cells to irinotecan,one of the three drugs of FOLFIRI,was resolved by treatment with mTORC1/2 inhibitors.Reitaet al[84]demonstrated that the combination of irinotecan and a mTORC1/2 blocker reduced migration and invasionin vitroas well as the development of liver metastasesin vivomore effectively than irinotecan alone.Consistently,the knockdown of Rictor increased the sensitivity to irinotecan in SMAD4-negative colon cancer cells[85].
Breast cancer liver metastasis
Breast cancer accounts for almost one in four cancer cases among women,thereby representing the leading cause of cancer in over 100 countries worldwide[80].Maet al[86]recently reviewed the evidence that after the bony skeleton and the lung,breast cancer metastasizes most often to the liver,leading to very limited survival if untreated.Although many systemic therapies exist for metastatic breast cancer,overactive PI3K/AKT/mTOR signaling was shown to be associated with resistance to therapy and with tumor progression[87-89].The findings revealed 92% Rictor positivity in breast cancer lymph node metastases[90]as well as decreased tumor growth and migration but increased apoptosis upon Rictor knockdown[91].Functionally,different pathways of mTORC2/Rictor involvement in breast cancer metastasis have been described.mTORC2 activates Rac1 through AKT phosphorylation and PKC-dependent downregulation of RhoGD12 leading to increased invasion and migration[46].Rac1 is also activated by IBP,which was shown to regulate migration and invasion as well as actin cytoskeleton rearrangement and matrix metalloprotease production in breast cancer cells[92]by activation of the mTORC2/AKT/FoxO3a signaling pathway[93].Moreover,interactions between mTORC2 and PRICKLE1 were shown to control cancer cell dissemination and motility[94].Similarly,Rictor interacts with PKC-ζ to regulate breast cancer metastasis[90].mTORC2 is further implicated in EMT in breast cancer by regulation of Snail and TGFß to control migration and invasion[21,95].
Melanoma liver metastasis
Melanoma liver metastasis occurs in up to 20% of patients with cutaneous melanoma and is one of the main prognostic factors of poor survival[96,97].mTORC2/Rictor is not only involved in PI3K dependent melanoma development[98]and metabolic reprogramming[99]but also in melanoma liver metastases.Rictor mRNA and protein were shown to be overexpressed in invasive melanoma[100]and to be significantly enhanced in metastatic compared with nonmetastatic melanoma[101].Consistent with those findings,siRNA-mediated Rictor knockdown as well as pharmacological inhibition of mTORC2 not only led to reduced tumor cell motility,migration,and invasionin vitro[100,101]but also reduced melanoma liver metastasisin vivo[101,102].Rictor depletion was shown to reduce AKT phosphorylation at the Ser473 and Thr308 residues and to inhibit the expression of MMP-2 and MMP-9[100,102].Moreover,upon Rictor inhibition,interaction with stromal components such as hepatic stellate cells and HGF-induced melanoma cell activation/motility was impaired[101].
Renal cancer liver metastasis
Liver metastases occur in about one-fifth of patients with metastatic renal cancer[103],and surgical therapy remains the only strategy to improve survival(see Pinottiet al[104]for review).However,mTORC2 signaling might be a potential therapeutic target,as it is involved in the formation of renal cancer liver metastasis.Sunet al[105]showed that the proinflammatory cytokines TNFα and IL-6 increased upregulation of Rictor through the NF-κB pathway,thereby enhancing chemotaxis,invasion,and migration of renal cancer cells.Upon Rictor knockdown,the formation of renal cancer liver and lung metastases was significantly reduced[105].Increased migration and invasion were also associated with activation of mTORC2/Akt/GSK3β/β-catenin signaling through TCTP overexpression[106].Furthermore,pharmacological mTORC2 inhibition led to reduced migration by regulation of HIF2α and increase of cell-celljunctionsviaE-cadherin[107].
Gastric and pancreatic cancer liver metastasis
Gastric cancer is the third leading cause of cancer-related deaths worldwide[80]with the liver being the most common side of gastric cancer metastasis[108].Similarly,pancreatic cancer is one of the most fatal diseases with a 5-year survival rate of only 7%[109].Rictor expression in gastric tumor samples was shown to correlate with TNM stage,lymph node metastasis,and poor long-term outcome.Positive staining of Akt at the Ser473 residue was associated with distant metastasis[7,110].Also,Wanget al[111]reported the role of mTORC2 in gastric cancer metastasis,as DDR2 was found to enhance invasion and EMT through mTORC2 activation and AKT phosphorylation.Upon Rictor knockdown,proliferation,migration,and invasion of gastric cancer cells were significantly reduced while apoptosis was enhanced[110].Regarding pancreatic cancer,Rictor protein expression was associated with overall survival after surgical resection.Patients with high or medium Rictor expression had significantly shorter survival compared with those with low expression[9].Upon siRNA-mediated Rictor knockdown,pancreatic tumor cell proliferation and vascularization were significantly impaired and a trend toward fewer liver metastases was observed[9].
CONCLUSION
Compared with mTORC1,little is known about the role of mTORC2 and its distinct subunit Rictor,in cancer.However,the present review underlines the importance and high relevance of mTORC2 not only in tumorigenesis of primary liver cancer but also in the formation of metastatic liver lesions with different primaries.Thereby,mTORC2/Rictor and AKT,its main downstream effector,are associated with various steps of the metastatic cascade,including EMT,migration and invasion,and angiogenesis,and tumor cell proliferation through different signaling pathways.However,a more refined understanding of the implications of mTORC2 in primary and secondary liver cancer is essential to convert this knowledge into the development of specific mTORC2 targeting therapies.
 World Journal of Gastrointestinal Oncology2021年11期
World Journal of Gastrointestinal Oncology2021年11期
- World Journal of Gastrointestinal Oncology的其它文章
- Hepatocellular carcinoma biomarkers,an imminent need
- Anatomical vs nonanatomical liver resection for solitary hepatocellular carcinoma:A systematic review and meta-analysis
- Atezolizumab plus bevacizumab versus sorafenib or atezolizumab alone for unresectable hepatocellular carcinoma:A systematic review
- Cell-free DNA liquid biopsy for early detection of gastrointestinal cancers:A systematic review
- Colorectal cancer in Arab world:A systematic review
- Induction chemotherapy with albumin-bound paclitaxel plus lobaplatin followed by concurrent radiochemotherapy for locally advanced esophageal cancer