血小板P选择素及CD40/CD40L在动脉粥样硬化中的研究进展
2021-11-13 07:32原达朱国斌
心血管病学进展 2021年10期
原达 朱国斌
(1.山西医科大学研究生院,山西 太原 030001; 2.山西医科大学附属第二临床医学院心内科,山西 太原 030001)
大量研究证实,炎症反应与动脉粥样硬化(atherosclerosis,AS)的发生、发展及其并发症密切相关。血小板具有血栓形成和免疫调节的双重作用,其活化后通过表面膜受体的表达及脱颗粒作用,释放多种活性物质,广泛参与AS的形成和易破裂斑块的发展[1-2]。血小板活化后释放趋化因子CCL5、单核细胞趋化因子-1(MCP-1,又名CCL2)和血小板因子4刺激白细胞募集,同时分泌炎症介质如可溶性CD40配体(sCD40L)、5-羟色胺、白介素(IL)-1β激活内皮细胞(endothelial cells,ECs)和白细胞,另一方面活化后的血小板膜表面表达P选择素(P-selectin,P-sel)、糖蛋白(GPⅠbα、GPⅥ和GPⅡb/Ⅲa)、Tool样受体2/4以及共刺激分子CD40L,介导血小板-白细胞和血小板-ECs的相互作用,促进炎症反应。血小板膜表面P-sel、CD40L分别与白细胞上的P选择素糖蛋白配体1(PSGL-1)、CD40结合后形成血小板-白细胞聚集体(platelet-leukocyte aggregates,PLA),增强炎症反应,活化ECs,促进白细胞招募、活化,加速AS的形成[3-4]。现对血小板P-sel和CD40/CD40L在AS中的炎症作用进一步描述与概括。
1 血小板P-sel
P-sel,又称CD62P,是SELP基因编码的,表达于活化血小板及ECs表面的跨膜糖蛋白,属于黏附分子选择素超家族成员。静息情况下,P-sel主要储存在血小板α颗粒和ECs的Weibel-Palade小体(Weibel-Palade body,WPB)中,细胞膜表面不表达或低表达,炎症损伤或激动剂作用下血小板活化脱颗粒仅数分钟便可高表达于膜表面或以可溶性P选择素(soluble P-selectin,sP-sel)脱落[4]。
1.1 P-sel与AS
在AS及炎症中,ECs表面P-sel与白细胞PSGL-1结合在白细胞的初始黏附中发挥直接作用,而血小板P-sel介导PLA形成,可加速白细胞在受损或炎症ECs上血小板沉积部位的初始黏附[4],另外PLA小体可诱导ECs中WPB的释放,导致ECs上P-sel增多。研究发现相比于ECs的P-sel,血小板P-sel在AS的发展和加速中发挥更为重要的作用。Burger等[5]对6周大小的载脂蛋白E基因敲除(ApoE-/-)Selp-/-和ApoE-/-Selp+/+小鼠骨髓行致死剂量辐射后移植重组获得四组基因型动物,36周后观察发现ECs表达P-sel的小鼠有较大的病变,ECs及血小板同时表达P-sel的小鼠比只有ECs表达P-sel的小鼠病变大30%,且钙化率更高。Zhang等[6]发现转人SELP基因的ApoE-/-小鼠(ApoE-/-Selp-/-TgSELP-/+)的主动脉斑块病变比基因敲除鼠Selp的ApoE-/-小鼠(ApoE-/-Selp-/-)更大,骨髓重组并在西式饮食15周后发现,ECs和血小板同时表达P-sel小鼠的主动脉斑块明显大于其他三组,其他三组则表现为斑块大小相等。此外,相较ECs的P-sel,血小板P-sel在动脉损伤后新内膜的形成中发挥主要作用,针对白细胞-血小板的治疗策略可有效抑制再狭窄。
1.2 血小板P-sel与AS
在AS中,血小板P-sel与白细胞PSGL-1结合形成PLA小体,一方面激活白细胞、ECs,上调细胞表面黏附分子表达,加强白细胞滚动,另一方面释放趋化因子、细胞因子等产物,诱导单核细胞趋化至血管壁炎症部位,并分化为巨噬细胞,促进AS形成[7]。Huo等[8]的研究首次阐明血小板P-sel在AS形成中的直接作用,向ApoE-/-小鼠多次注射表达P-sel的血小板,观察到PLA形成的同时血小板源性趋化因子CCL5、CXCL4向白细胞、ECs表面传递,同时有更多的白细胞与血管细胞黏附分子-1结合,这表明活化血小板通过表达P-sel介导形成PLA的同时向单核细胞/白细胞和血管壁传递血小板源性趋化因子及促炎因子,参与AS的形成。另外,PLA中血小板亦可传递血小板源髓样相关蛋白-14(MRP-14)至单核细胞,研究发现外周AS疾病患者血小板MRP-14及其mRNA显著增多,且与PLA呈正相关,并于电镜下观测到PLA小体的单核细胞中有免疫荧光标记的血小板源MRP-14,靶向抑制血小板MRP-14可下调单核细胞肿瘤坏死因子(tumor necrosis factor,TNF)-α、IL-1β和CCL2的表达;进一步用重组MRP-14刺激血小板后发现MRP-14可剂量依赖性地增加血小板P-sel的表达[9]。可见,血小板MRP-14可通过增加P-sel的表达导致单核细胞-血小板聚集物的形成,同时靶向血小板MRP-14可为AS的抗炎治疗提供新思路。
在血栓形成中,血小板P-sel主要介导血小板在ECs表面的初始“滚动”,GPⅠb、GPⅡb/Ⅲa主要在血小板的稳定黏附中起作用[4],但P-sel对GPⅡb/Ⅲa-纤维蛋白原的结合有稳固作用,有利于形成更大的血小板聚集体[10]。使用P-sel单抗可抑制剪切力诱导的血小板聚集,并对GPⅡb/Ⅲa单抗的抑制有叠加作用。此外,血小板P-sel与白细胞结合后亦可发挥促凝作用,可诱导白细胞释放组织因子、白细胞微粒及炎症因子激活凝血途径,促进纤维蛋白原沉积和血栓形成[11]。
此外,中性粒细胞胞外陷阱(neutrophil extracellular trap,NET)在AS和血栓形成中发挥重要作用,阻断P-sel与PSGL-1相互作用可抑制由凝血酶激活的血小板与中性粒细胞结合引起的NETosis(NET的形成过程),这一途径可能是治疗NET相关AS疾病的潜在治疗靶点[12-13]。
2 血小板CD40/CD40L
CD40L及其受体CD40属于TNF和TNF受体家族的一对共刺激分子,在免疫调节和炎性反应中发挥关键作用。CD40L是一种Ⅱ型包膜糖蛋白,主要表达于活化的CD4+T细胞,在血小板上也有表达,其在细胞外区域包含一个保守的TNF同源结构域,可实现三聚化并和受体结合,CD40主要表达于抗原提呈细胞及血管壁细胞表面,此外血小板表面亦有CD40表达。CD40L储存在α颗粒中,血小板活化后迅速释放到膜表面,被细胞外蛋白酶剪切下来形成sCD40L,循环中>95%的sCD40L来自血小板[14-15]。
2.1 CD40/CD40L与AS
在单核细胞中,CD40-TRAF6通路发挥促AS作用,而CD40-TRAF2/3/5通路主要维持正常免疫功能。动物实验发现,基因敲除CD40或抗体抑制CD40L后,可在有AS倾向的低密度脂蛋白受体基因敲除(LDLR-/-)或ApoE-/-小鼠中形成炎症程度较低的较小AS病变。CD40与肿瘤坏死因子受体相关因子(TRAF)1/2/3/5/6结合后启动不同的调节因子和效应因子,导致斑块向有利表型转化主要依赖于阻断CD40-TRAF6通路,Lutgens等[16]发现白细胞中缺乏CD40-TRAF6通路的ApoE-/-小鼠斑块仅表现为少量的脂质条纹及少量的白细胞浸润和基质金属蛋白酶2/9,外周白细胞特别是Ly6C+单核细胞的数量及黏附功能均下降,其中Ly6C+单核细胞属于促炎效应细胞,较易进入动脉壁并分化为巨噬细胞;少数浸润动脉壁的单核细胞分化为产生IL-10的抗炎M2型巨噬细胞,且细胞内仅含少量核因子κB抑制蛋白α(IκBα)。这表明阻断CD40-TRAF6通路的抗AS作用与巨噬细胞分化为抗炎M2表型及抑制核因子κB(NF-κB)信号有关。目前认为CD40-TRAF2/3/5通路是维持正常免疫所必需的,在AS中发挥平衡的作用[16-17],而单独的CD40-TRAF5通路具有抗AS作用(图1A)[18]。另有学者认为CD40L诱导的AS与CD40无关,主要通过与巨噬细胞分化抗原-1(Mac-1)作用引起[19]。抑制CD40L-Mac-1相互作用不仅抑制AS的炎症反应,同时可避免完全抑制CD40L后带来的免疫抑制和血栓栓塞效应[20]。
2.2 血小板CD40/CD40L与AS
在血小板中,CD40L和CD40均有促AS作用,另外sCD40L可与血小板上CD40、GPⅡb/Ⅲa和α5β1结合促血栓形成。给ApoE-/-小鼠注射缺乏CD40L或CD40的血小板,不仅斑块明显减小且斑块内巨噬细胞浸润、凋亡及斑块内出血显著减少[21-22]。血小板CD40L通过诱导ECs分泌趋化因子IL-8、CCL2,并使其表达黏附分子E选择素、血管细胞黏附分子-1和细胞间黏附分子1,增强白细胞、血小板对ECs的黏附力;另一方面,CD40L介导PLA的形成,加速白细胞活化并通过CCL2募集至血管壁,加速AS[21]。同样,血小板CD40通过激活ECs表达血管细胞黏附分子-1、血小板内皮细胞黏附分子-1、血管内皮细胞钙黏蛋白及P-sel,促进白细胞的黏附、活化及分泌IL-1β,加强血管壁炎症反应[22]。血小板CD40L及CD40在PLA的形成中均发挥重要作用。Lievens等[21]研究发现当血小板缺乏CD40L或白细胞缺乏CD40时,PLA形成显著减少,同时在胶原上血小板CD40L缺陷的血栓上P-sel暴露减少,并推测CD40/CD40L相互作用形成PLA可能是血小板P-sel依赖的。Gerdes等[22]的研究则证明血小板CD40也是通过间接影响血小板P-sel的表达介导PLA的形成,这与Jin等[23]的研究一致。此外血小板CD40L通过影响T细胞亚群分布干扰效应T细胞/调节性T细胞平衡来加速AS病变,用CD25耗尽注射缺乏CD40L血小板的ApoE-/-小鼠调节性T细胞后,则其抗AS作用消失[21]。
在AS中血小板CD40L依赖的血栓形成主要在胶原暴露的晚期发挥作用,缺乏CD40L的ApoE-/-小鼠血小板在胶原上的聚集明显受损,而在血管性血友病因子的黏附与CD40L+/+ApoE-/-小鼠表现相似[21]。CD40L通过血小板CD40可引起α颗粒和致密颗粒释放,导致P-sel、GPⅡb/Ⅲa表达增加以及典型的形态学改变。CD40L可通过CD40依赖的不同途径激活血小板,一方面通过CD40/TRAF2/Rac1/p38丝裂原活化蛋白激酶(MAPK)信号通路加剧血小板活化和聚集,突变形式的sCD40L和CD40-/-小鼠血小板无法引起这种反应,另一方面sCD40L亦可触发血小板中NF-κB的强烈激活,抑制IκBα可逆转CD40L诱导的IκBα磷酸化,但不影响p38 MAPK磷酸化,同样抑制p38 MAPK对IκBa磷酸化也无影响[24-25]。Kojok等[26]的研究结果则证明CD40L激活血小板NF-κB是CD40依赖性的,通过CD40L/CD40/TAK1/NF-κB信号启动血小板并增强血小板的聚集,阻断血小板膜上的另外两种可与CD40L结合的受体α5β1或GPⅡb/Ⅲa后,对IκBα的磷酸化并无影响。此外,在高剪切应力下,CD40L可通过KGD整合素序列直接与血小板GPⅡb/Ⅲa结合,增强血小板的聚集和血栓的稳定性[27]。Kuijpers等[28]通过体外刺激ApoE-/-小鼠血小板胶原受体GPⅥ后发现,与缺乏CD40的血小板相比,缺乏CD40L会引起GPⅡb/Ⅲa的激活和P-sel表达减少,进一步用CD40L刺激血小板,在缺乏CD40L和CD40血小板中,均可引起PI3K-Akt信号通路中胶原诱导的Akt磷酸化增强以及更大的血小板聚集体形成,基因缺失或抑制PI3K-β可抑制这一增强现象,而靶向NF-κB酶抑制剂α(IKKα)阻断非经典NF-κB通路并未减弱CD40L增强血小板聚集的作用;尽管靶向IKKβ和IκBα阻断经典NF-κB通路表现出对血小板聚集的抑制作用,但该学者认为添加抑制剂会影响血小板的Ca2+信号从而减弱血小板聚集。这表明CD40L在AS稳定血栓的形成作用中,至少部分是依赖于GPⅡb/Ⅲa的激活而与CD40无关(图1B)。此外CD40L可与血小板表面整合素α5β1结合导致血小板活化,促进血栓形成[29]。CD40L可同时与CD40和α5β1结合形成CD40L-CD40-整合素复合物,在CD40/CD40L信号通路中发挥关键作用,缺乏整合素结合位点的CD40L突变体可拮抗CD40/CD40L相互作用[30]。
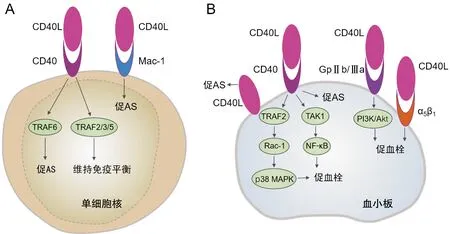
图1 A:CD40L在单核细胞中的促AS通路;
3 诊断与治疗
3.1 sP-sel
sP-sel不仅是血小板活化及血栓的标志物,也是炎症的标志物,大量的临床研究表明sP-sel在AS及炎症性疾病中处于高表达状态。近期的一项单中心前瞻性队列研究发现经皮冠状动脉介入治疗(percutaneous coronary intervention,PCI)前sP-sel浓度是术后双联抗血小板治疗(阿司匹林联合氯吡格雷)期间高残余血小板反应性的独立危险因素,而PCI后血小板高反应性患者有更高的心血管不良事件及支架内血栓发生[31-32]。
3.2 sCD40L
sCD40L水平在冠心病危险因素吸烟、高血压、糖尿病、高胆固醇血症、高同型半胱氨酸血症、不稳定型心绞痛和急性心肌梗死的患者中均升高,同时也可用来预测急性冠状动脉综合征的未来不良事件。此外,他汀类药物、抗高血压药和抗血小板药,已被认为是sCD40L水平的潜在调节剂,对急性冠状动脉综合征患者的预后有积极影响[33]。早期的体外试验发现sCD40L可降低ECs一氧化氮水平,导致内皮依赖性血管舒张功能障碍,近期Aslan等[34]在一项152例冠状动脉微血管疾病患者的研究中,发现与对照组相比,患者血清sCD40L水平与超敏C反应蛋白呈正相关,推测冠状动脉微血管疾病患者血清中较高的sCD40L浓度可能与炎症激活有关。目前认为冠状动脉微血管疾病是心肌缺血的三种经典机制之一,也有学者认为是AS累及冠状小、微动脉的一种早期病变。Gergei等[35]研究了sCD40L作为心血管全因死亡率标志物的潜在作用,发现只在冠状动脉疾病患者和射血分数保留性心力衰竭的亚组分析中,才能显示sCD40L水平与心血管短期死亡率之间的显著相关性。
3.3 治疗
最近的一项临床试验发现使用P-sle单抗inclacumab可显著降低非ST段抬高型心肌梗死患者PCI后的肌钙蛋白I和肌酸激酶水平,且在PCI前3 h给药可获得最佳效果[36],这为通过靶向P-sel来减少心肌损伤提供了强有力证据,但仍需大量临床数据支持。CD40/CD40L相互作用在免疫调节和血栓稳定性中发挥重要作用,单纯地阻断CD40L或CD40会导致血栓栓塞和免疫抑制事件发生,有学者将TRAF6抑制剂合并到重组高密度脂蛋白纳米颗粒中靶向于单核/巨噬细胞可降低AS的发生,在ApoE-/-小鼠和灵长类动物中均未观察到活性纳米颗粒的毒性作用[17]。此外,血小板CD40在动脉损伤后新生内膜形成中起关键作用,可作为预防PCI后再狭窄的治疗靶点[23]。
4 展望及总结
AS是一种炎性疾病,TNF-α驱动的炎症过程在其中发挥重要作用,研究发现,在衰老相关炎症中,TNF-α通过对巨核细胞转录重编辑的调控和血小板线粒体功能的影响,促进血小板反应性增高和血栓形成,可见血小板在AS炎症中的重要角色[37]。血小板是联系炎症、免疫和血栓三者间的桥梁,血小板P-sel和CD40/CD40L在AS形成及血栓形成中发挥重要作用,同时循环中sP-sel、sCD40L可作为AS的生物标志物。目前的研究主要集中在ECs的P-sel和白细胞CD40/CD40L以及相关信号通路,且靶向治疗也仅在动物实验和小规模临床试验中得到验证。对于血小板P-sel和CD40/CD40L及它们与白细胞、ECs之间的关系在AS疾病中的充分研究,不仅有益于降低炎症和血栓残余风险,同样也可为预防抗炎治疗所带来的免疫抑制和血栓栓塞等并发症提供帮助。
猜你喜欢
武汉大学学报(医学版)(2022年2期)2022-11-23
材料与冶金学报(2022年2期)2022-08-10
北华大学学报(自然科学版)(2022年3期)2022-07-11
当代陕西(2022年5期)2022-04-19
中国宝玉石(2021年5期)2021-11-18
医学食疗与健康(2021年25期)2021-05-12
科学与财富(2021年33期)2021-05-10
现代装饰(2021年1期)2021-03-29
科学导报(2020年25期)2020-04-28
人人健康(2017年19期)2017-10-20