
Phylogenetic status of the cicada tribe Sinosenini (Hemiptera:Cicadidae) based on mitochondrial genomes
2021-11-06 08:04YUANFeiMinWEICong
昆虫学报 2021年10期
YUAN Fei-Min, WEI Cong
(Key Laboratory of Plant Protection Resources and Pest Management of the Ministry of Education, College of Plant Protection,Northwest A&F University, Yangling, Shaanxi 712100, China)
Abstract: 【Aim】 The aim of this study is to clarify the phylogenetic status of the tribe Sinosenini in Cicadoidea, in which the cicadas have no timbal organs. 【Methods】 Based on adult specimens of Karenia caelatata of Sinosenini collected in Ningshan, Shaanxi, northwestern China, the mitochondrial genome of K. caelatata was sequenced, annotated, bioinformatically analyzed, and compared with the mitochondrial genomes of other taxa of Cicadoidea. Molecular phylogenetic trees for Cicadoidea were constructed using both maximum likelihood (ML) method and Bayesian inference (BI) method. 【Results】 The mitochondrial genome of K. caelatata (GenBank accession number: MN922304) is 14 960 bp in length, and the gene organization, nucleotide composition and codon usage of protein-coding genes display similar structural characteristics to those of other taxa of Cicadoidea. Analysis of nucleotide diversity reveals that genes atp8, nad6 and nad2 are highly variable, while cox1 is more conserved. Ratios of nonsynonymous and synonymous substitution rates indicate that the evolution of mitochondrial genomes in Cicadoidea is under a high-level purifying selection. Results of phylogenetic analyses support the monophyly of Cicadomorpha, revealing the relationship of the three superfamilies of this infraorder as (Membraciodea+(Cicadoidea+Cercopodidea)). Karenia is clustered with representatives of the tribe Dundubiini in the subfamily Cicadinae and most closely related to the genus Meimuna. Mogannia and Vagitanus, representatives of Cicadatrini, are clustered with the members of Cicadettinae. Yezoterpnosia is not a monophyletic group. 【Conclusion】 Sinosenini should be transferred from Cicadettinae to Cicadinae and merged with Dundubiini, while Cicadatrini should be transferred from Cicadinae to Cicadettinae. Our results provide new information for future study of evolution of cicadas with different sound-producing mechanisms.
Key words: Cicadoidea; Cicadomorpha; timbal organ; mitogenome; nucleotide diversity; evolutionary rate
1 INTRODUCTION
Insect mitochondrial genomes (mitogenomes) are typically closed, circular, double-stranded DNA molecules with relatively conserved gene organization, order and direction, ranging from 14 to 20 kb in length. Each insect mitogenome is composed of a typical set of 37 genes including 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes, and a large non-coding region or A+T-rich region (also referred to as the control region, CR) (Wolstenholme, 1992a; Boore, 1999; Cameron, 2014). In comparison with individual mitochondrial or nuclear genes, mitogenomes have unique features including high copy numbers, rare recombination, relatively high evolutionary rates, maternal inheritance and no introns, which can provide genome-level information like nucleotide composition, structural genomic features, and gene rearrangement (Curole and Kocher, 1999; Lin and Danforth, 2004). Mitogenomes have been widely used in multiple research areas such as species delimitation, population genetics, phylogenetic analyses, and comparative genomics at different taxonomic levels (Songetal., 2017; Johnsonetal., 2018;ukasiketal., 2019). Along with the rapid progress of high-throughput sequencing and DNA analysis technologies, a good deal of insect mitogenomes have been sequenced and placed in public database. Mitogenome data have been commonly used to investigate the phylogenetic relationships among taxa in Hemiptera at various hierarchical levels (Dietrichetal., 2017; Lietal., 2017; Voronovaetal., 2020). For all that, the phylogenetic status of some taxa within Hemiptera inferred based on extant morphological and/or molecular data remains controversial due to the fact that mitogenome data of some taxa are unavailable.
Cicadas are well known for their ability to produce loud sounds by males. Currently, the Cicadoidea is composed of Tettigarctidae and Cicadidae, including ~3 100 known species worldwide (Sanborn, 2013). The Cicadidae comprises all species of cicadas, except the two extant species of the sister-family Tettigarctidae distributed in Australia. The main method of sound production in cicadas is the timbal mechanism, and the higher classification of Cicadoidea mainly based on characteristics of the sound organs (i.e., timbals) and related morphological structures (Moulds, 2005). However, not all cicadas produce sound using the timbals, and species of some genera,i.e.,KareniaDistant,LamotialnaBoulard,PlatypediaUhler,NeoplatypediaDavis andMaroboduusDistant (=YdiellaBoulard), lack the timbal organs. These five genera were originally placed in Tibicinidae by Distant (Distant, 1906), and then transferred from Tibicinidae to Platypediidae (Boulard, 1973, 1976). However, Boulard (1988) disbanded Platypediidae and distributed these five genera to three different subfamilies of Tibicinidae withKareniadistributed in Tibicininae, since they probably lost the timbal organs independently. Chouetal.(1997) describedKareniasulcataLeietChou, but they followed Boulard’s original concept of Platypediidae by includingKarenia, though they treated it as a subfamily rather than a family of Cicadidae. Moulds (2005) conducted morphological phylogenetic analyses of Cicadoidea, and divided Cicadidae into three subfamilies, in whichKareniawas included in Cicadettinae (=Tibicininaeauct.). However, the phylogenetic status of this group has not yet been solved, because no taxa ofKareniawere included in the phylogenetic analyses of Moulds (2005). Recently, the results of a phylogenetic study by Marshalletal. (2018) indicated thatKareniais a member of Cicadinae, but the exact placement was poorly supported. More recently,ukasiketal. (2019) and Simonetal. (2019) conducted phylogenetic analyses for Cicadidae, which indicated thatPlatypediabelongs to Tibicininae, but no other taxa that lack timbal organs were included in their analyses.
In the present study, we sequenced and analyzed the mitogenome ofK.caelatatausing next-generation sequencing (NGS), and performed phylogenetic analyses of Cicadoidea based on mitogenomic data. The purpose of this research is: 1) to provide a detailed comparative analysis of Cicadoidea mitogenomes, including gene order, nucleotide composition, composition biases, codon usage and tRNA secondary structures; and 2) to reveal the phylogenetic relationships within Cicadoidea, with special reference to the systematic status ofKarenia(Sinosenini). The results will provide useful information for future phylogenetic research of the Cicadoidea.
2 MATERIALS AND METHODS
2.1 Sample collection and DNA extraction
Adults ofK.caelatatawere collected in August, 2018 at Huoditang Forestry Farm in Ningshan County, Shaanxi Province, northwestern China (33.43°N, 108.45°E). All fresh adult specimens were captured by light trapping and immediately preserved in 100% ethanol, then stored at -80℃ in the laboratory at the Entomological Museum of Northwest A&F University (NWAFU), Yangling, Shaanxi Province, China. Total DNA was extracted from the thoracic muscles using the DNeasy DNA Extraction Kit (Qiagen) following the manufacturer’s instructions. The NanoDrop 2000 spectrophotometer was utilized to quantify the extracted DNA for library construction. Voucher specimens are deposited at NWAFU. Insects used in this study are not endangered or protected species, and no specific permits were required for collection.
2.2 Sequencing, assembly and annotation
The whole mitogenome ofK.caelatatawas sequenced by NGS on an Illumina HiSeqTM2500 platform with paired-ends of 2×150 bp (Biomarker Technologies Corporation, Beijing). Raw paired reads were retrieved and quality-trimmed with default parameters, and clean reads were preliminarily assembled usingdenovoassembly with the minimum contig length >8 000 bp in the CLC Genomics Workbench v11.0 (https:∥www.qiagenbioinformatics.com/)(CLC Bio, Aarhus, Denmark). The reads were assembled into the circular mitogenome in Geneious v10.0.5 (Biomatters Ltd, Auckland, New Zealand) (Kearseetal., 2012), which employed the mitogenome ofMeimunaopalifera(Cicadidae: GenBank: KY039112) as a bait sequence (Songetal., 2017). The mitogenome ofK.caelatatawas annotated with Geneious v10.0.5, also withM.opaliferaas the reference. All the 13 PCGs were predicted by the ORF Finder in NCBI (https:∥www.ncbi.nlm.nih.gov/orffinder) employing the invertebrate mitochondrial genetic codes and modified by comparison with the previously published mitogenomes of Cicadoidea from GenBank. The two rRNA genes (rrnSandrrnL) and control region (A+T-rich) were identified based on the locations of adjacent genes and by comparison with the homologous sequences from other species within Cicadoidea. The 22 tRNA genes were identified with the invertebrate mitochondrial genetic codes using Mitos Web Server (http:∥mitos.bioinf.uni-leipzig.de/index.py) (Berntetal., 2013) and tRNAscan-SE Server (http:∥lowelab.ucsc.edu/tRNAscan-SE/) (Lowe and Eddy 1997). The clover-leaf secondary structures were predicted by the Mitos Web Server, and the secondary structures were manually plotted with Adobe Illustrator CC2017 according to the results of the predictions. The physical mitogenomic circular map was drawn using Organellar Genome Draw (OGDRAW) (https:∥chlorobox.mpimp-golm.mpg.de/OGDraw.html) (Lohseetal., 2013).
2.3 Bioinformatic analysis
The nucleotide composition, codon usage and relative synonymous codon usage (RSCU) values of each PCG ofK.caelatatawere calculated using Phylosuite v1.1.16 (Zhangetal., 2020). Strand asymmetry was calculated using the following formulas: AT-skew=[A-T]/[A+T] and GC-skew=[G-C]/[G+C] (Perna and Kocher, 1995). The tandem repeats of the control region were identified by the Tandem Repeat Finder Online Server (http:∥tandem.bu.edu/trf/trf.html) (Benson, 1999), and the stem-loop structure was inferred by Mfold Web Server (http:∥mfold.rna.albany.edu/). Gene arrangements were investigated by comparing the newly sequenced mitogenome with all the previously sequenced mitogenomes of Cicadoidea available from GenBank. A sliding window of 200 bp in a step size of 20 bp was used to estimate nucleotide diversity (Pi) with Dnasp v6 (Rozasetal., 2017) among 13 PCGs and 2 rRNA genes across the alignment from the mitogenomes of 63 species of Cicadoidea. The ratio of non-synonymous (Ka) and synonymous (Ks) substitution rates for each PCG was calculated with Dnasp v6.
2.4 Phylogenetic analysis
Phylogenetic analyses were performed based on the 13 PCGs and two rRNA genes of the mitogenomes of 71 species which are currently available, including 69 species of Cicadomorpha (62 species of Cicadidae, one species of Tettigarctidae, two species of Membracidae, two species of Cicadellidae, and two species of Cercopidae) as ingroups plus two species of Fulgoromorpha as outgroups (Songetal., 2012, 2017; Liuetal., 2014; Van Leuvenetal., 2014; Duetal., 2017a, 2017b; Lietal., 2017; Su and Liang, 2018; Duetal., 2019; Huetal., 2019;ukasiketal., 2019; Wangetal., 2019). With the exception of mitogenome data ofK.caelatata, the available mitogenomes of 61 cicada species were downloaded from GenBank (the accession numbers of all taxa are provided in the phylogenetic tree below), which belong to four of the five subfamilies of Cicadidae (no mitogenome of species of Tettigomyiiane has been sequenced yet).
Each PCG gene was individually aligned with MAFFT v7.313 using G-INS-I strategy in codon-alignment mode (Katoh and Standley, 2013). The two rRNA genes were aligned separately using the muscle algorithm implemented in MEGA v7.0.26 (Kumaretal., 2016), and unreliably aligned regions were removed using Gblocks v0.91b (Talavera and Castresana, 2007). The results were concatenated using Phylosuite v1.1.16 (Zhangetal., 2020). Potential substitution saturation of each dataset was assessed using the index of substitution saturation (Iss) of Xiaetal.(2003) implemented in the DAMBE 5 (Xia, 2013). The optimal nucleotide substitution models and partition strategies for maximum likelihood (ML) and Bayesian inference (BI) phylogenetic analyses were recommended using PartitionFinder v2 (Lanfearetal., 2017). The best-fitting model was selected for each partition with the “greedy” search algorithm, “linked” to estimate branch lengths using the Bayesian Information Criterion (BIC). Alignments of individual genes were concatenated to generate four datasets: 1) the PCG123 dataset, including all three codon positions of 13 PCGs; 2) the PCG123R dataset, including all three codon positions of 13 PCGs and two rRNA genes; 3) the PCG12 dataset, including the 1st and 2nd codon positions of 13 PCGs; and 4) the PCG12R dataset, including the 1st and 2nd codon positions of 13 PCGs and two rRNA genes.
The ML analyses were conducted using IQ-tree v1.6.8 (Nguyenetal., 2015), under the ultrafast bootstrap approximation approach (Minhetal., 2013) with 1 000 replicates. Additionally, the BI analyses were performed using MrBayes v3.2.6 (Ronquistetal., 2012) through the Cipres Science Gateway v3.3 (https:∥www.phylo.org/)(Miller, 2011). ML and BI trees constructed based on four datasets were drawn using iTOL v5.5.1 (https:∥itol.embl.de/)(Letunic and Bork, 2019).
3 RESULTS
3.1 General features of mitogenome organization and nucleotide composition of K. caelatata
The mitogenome ofK.caelatata(14 960 bp in length) is a closed, circular and double-stranded DNA molecule (Fig. 1) (GenBank accession number: MN922304), including a typical set of 37 insect mitochondrial genes (13 PCGs, 22 tRNA genes and 2 rRNA genes) and a large non-coding control region (A+T-rich region). Twenty-three of the 37 coding genes, including 9 PCGs and 14 tRNA genes, are located on the majority strand (J-strand), while the remaining 14 genes (4 PCGs, 8 tRNA genes and 2 rRNA genes) are located on the minority strand (N-strand) (Table 1). The gene organization ofK.caelatatamitogenome is consistent with other mitogenomes of Cicadoidea as well as that ofDrosophilamelanogaster(Roberti, 2003). The A+T content, accounting for 78.30% of the entire mitogenome ofK.caelatata, shows a strong AT nucleotide bias (A: 38%, T: 40.30%, G: 10.30%, C: 11.40%). This is an intermediate value to all reported mitogenomes of Cicadoidea, ranging from 72.30% (Mendozanaplatypleura) to 80.50% (Yezoterpnosiavacua). The mitogenome ofK.caelatataexhibits a negative AT-skew (-0.028) and GC-skew (-0.047) on the whole, indicating that T and C are more abundant than A and G.
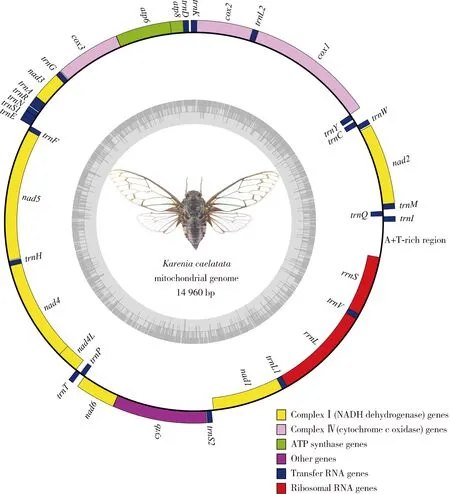
Fig. 1 Circular map of the mitochondrial genome of Karenia caelatataThe genes on the outer circle are located on the majority strand (J-strand) whereas the genes on the inner circle are located on the minority strand (N-strand). Protein-coding genes and ribosomal genes are shown with standard abbreviations.
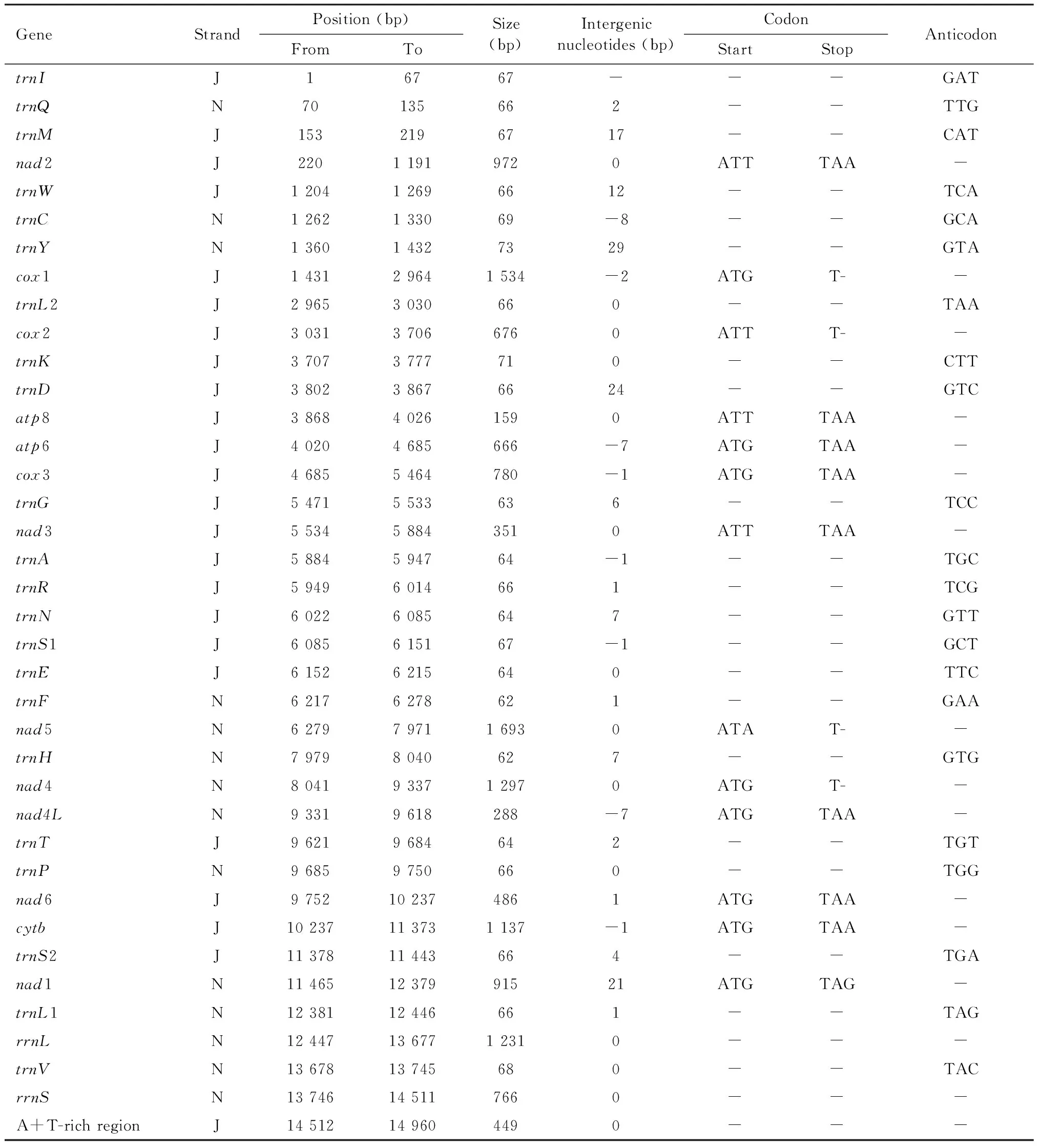
Table 1 Mitochondrial genome organization of Karenia caelatata
The mitogenome ofK.caelatatais relatively compact, with eight pairs of genes possessing overlapping regions (Table 1). The longest overlap istrnW-trnC(8 bp), followed byatp8-atp6 (7 bp),nad4-nad4L(7 bp),trnY-cox1 (2 bp),atp6-cox3 (1 bp),nad3-trnA(1 bp),trnN-trnS1 (1 bp), andnad6-cytb(1 bp). When the control region in the mitogenome ofK.caelatatawas excluded, a total of 15 intergenic spacers with 135 bp non-coding bases remained (Table 1), ranging from 1 bp to 29 bp. The longest intergenic spacer is located betweentrnCandtrnY(29 bp), followed bytrnK-trnD(24 bp),trnS2-nad1 (21 bp), andtrnQ-trnM(17 bp).
3.2 Protein-coding genes (PCGs) and codon usage of the mitogenome of K. caelatata
The mitogenome ofK.caelatataincludes the usual set of 13 PCGs with 10 950 bp in total length. Nine genes (nad2,cox1,cox2,atp8,atp6,cox3,nad3,nad6 andcytb) are located on the J-strand, while the others (nad1,nad5,nad4 andnad4L) are on the N-strand. The PCGs have the lowest A+T content (77.80%), and the 3rd codon position has a significantly higher A+T content, which is much higher than that of the 1st and 2nd positions (90.55% versus 74% and 68.80%). The A+T content and AT-skew of the entire protein-coding region are similar with other mitogenomes of Cicadoidea. All the 13 PCGs inK.caelatataare initiated strictly by a canonical start codon ATN (ATA/T/G) and ended with the stop codon TAA/G or its truncated form, a single T- (Table 1). This is the same with most species of Cicadoidea.
The RSCU values and the amino acid composition in the mitogenome ofK.caelatata(Fig. 2) and those of other cicadas indicate that the top five frequently utilized amino acids in the mitogenomes of all the 63 species of Cicadoidea are Ile, Leu, Met, Phe and Ser; and the most frequently used codons are AUU (Ile), UUA (Leu), UUU (Phe), AUA (Met) and AAU (Asn). All of them are composed solely of A or U. The amino acid codons ended with A/T are more frequent than those terminated with G/C, reflecting that a high A+T content occurred.
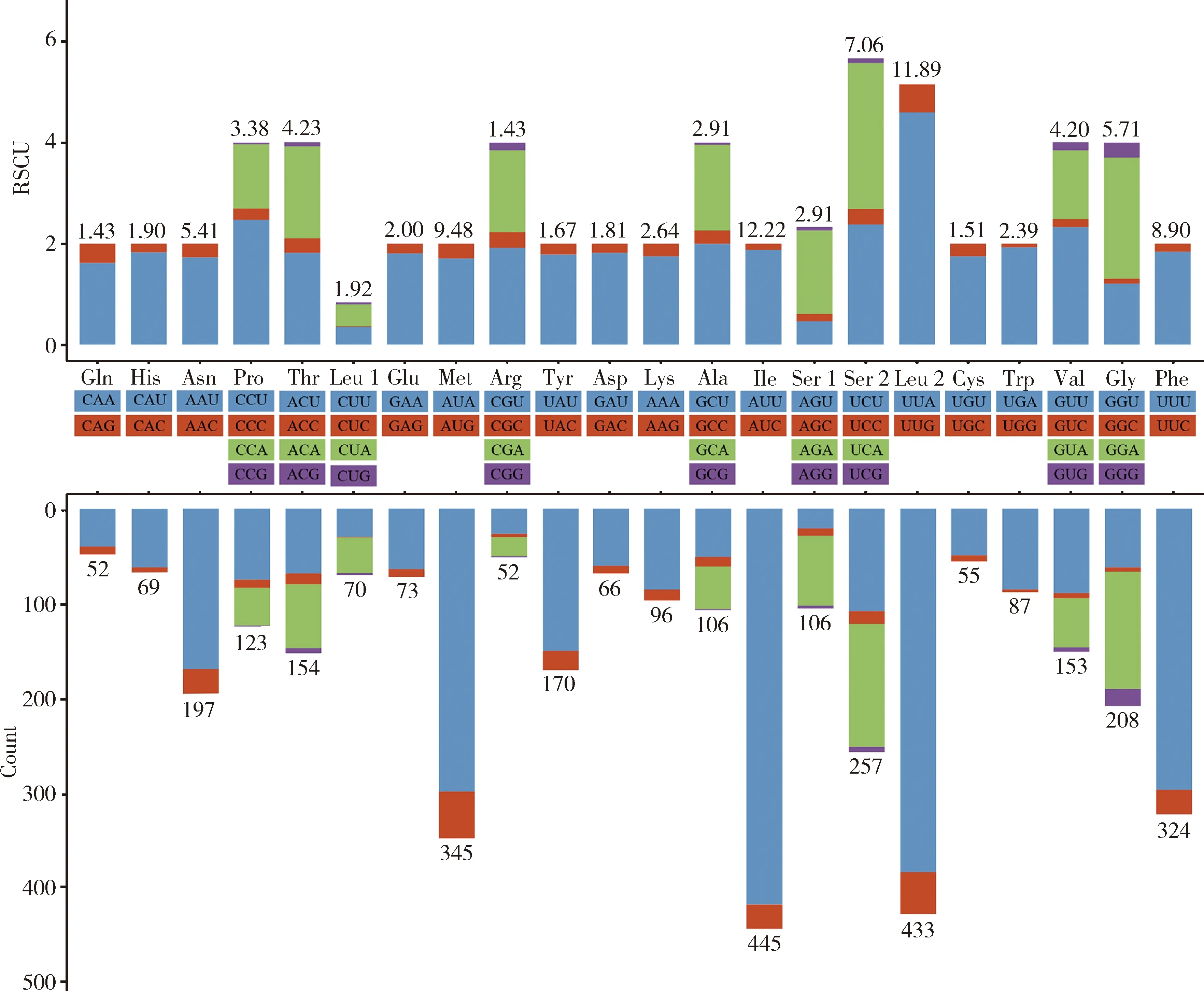
Fig. 2 Relative synonymous codon usage (RSCU) in 13 PCGs of the mitogenome of Karenia caelatataThe stop codon is not given. Codon families are displayed between the two X-axis.
3.3 tRNA genes and rRNA genes of the mitogenome of K. caelatata
The mitogenome ofK.caelatataincludes 22 canonical tRNA coding genes which are discontinuously scattered across the entire mitogenome (Fig. 1; Table 1). The total length of the 22 tRNA genes is 1 453 bp; the A+T content of tRNA genes is 81.35%, which is higher than that in other regions. The length of the 22 tRNA genes ranges from 62 bp (trnFandtrnH) to 73 bp (trnY) due to variation in the length of the variable loop (Table 1), which is similar to other mitogenomes within Cicadoidea. The anticodon nucleotides for the corresponding tRNAs are consistent with those presented in other cicada species. With the exception oftrnS, the remaining 21 tRNAs could be folded into the common clover-leaf secondary structures. WhiletrnS1 is an exception due to the lack of a dihydrouridine (DHU) arm, which forms a simple loop (Fig. 3).

Fig. 3 Inferred secondary structures of 22 tRNAs in the mitogenome of Karenia caelatataDashes indicate Watson-Crick bonds, and dots indicate an unmatched base pairing. The tRNAs are labeled with the abbreviations of their corresponding amino acids, standard abbreviations, and the IUPACIUB single-letter amino acid codes.
Considering the secondary structure of tRNAs from cicadas having never been predicted, the prediction of their secondary structure was performed in the present study (Fig. 3). Based on comparative analyses of the predicted secondary structures, besides the classic A-U and G-C pairs, four types of mismatches (i.e., G-U, U-U, A-A and U-C) in the amino acid acceptor arm, DHU arm, TψC arm and anticodon arm were found in 15 tRNAs (Fig. 3). There is an extra unpaired adenine in the anticodon arm oftrnS1 inK.caelatata. The generrnSis located betweentrnVand the A+T-rich region. The generrnLis located at a conserved position betweentrnL1 andtrnV(Fig. 1). These two rRNA genes are located on the N-strand, exhibiting positive GC-skew (0.222 and 0.188, respectively) and negative AT-skew (-0.057 and -0.026, respectively), which is consistent with that of other cicada species. The A+T content ofrrnLis higher thanrrnS(81% and 79.10%, respectively).
3.4 Control region of the mitogenome of K. caelatata
The putative control region (the major non-coding region or A+T-rich region), believed to be involved in regulating the transcription and replication of mitochondrial DNA in insects, is the longest intergenic spacer in the mitogenome (Zhangetal., 2019). It is located betweenrrnSandtrnIin the putative ancestral arrangement of insects, and is the most variable region in length and organization among the mitogenomes of Cicadoidea (Fig. 1). The control region of mitogenomes of sequenced cicadas varies from 575 bp (Platypleurakaempferi) to 829 bp (Pomponialinearis), but it is unavailable in some cicada species due to the complicated secondary structures and the high A+T content. The full length of the control region inK.caelatatamitogenome is 499 bp, with an A+T content of 78.30%, showing slightly negative AT-skew (-0.028) and GC-skew (-0.047). This is consistent with the whole mitogenome. No tandem repeat elements were found in the control region, but some stem-loop structures and subregions with high A+T content were found at the 3′ end of the control region.
3.5 Nucleotide diversity and evolutionary rate of the mitogenome of K. caelatata
The sliding window analyses of the aligned 13 PCGs and the two rRNA genes of the mitogenomes of the 63 species of Cicadoidea exhibit highly variable nucleotide diversity (Pi). The shortest PCG gene,atp8 (Pi=0.341), presents the highest variability, followed bynad6 (Pi=0.302) andnad2 (Pi=0.281). In contrast,cox1 (Pi=0.175),cox3 (Pi=0.202) andnad1 (Pi=0.205) show relatively low Pi values, indicating that they are relatively conserved genes among the 13 PCGs. The two rRNA genes are also highly conserved, with lower Pi values of 0.192 inrrnSand 0.195 inrrnL, respectively.
To assess the evolutionary rate of the 13 PCGs of the mitogenome ofK.caelatata, the values of Ka and Ks were calculated for each PCG, which can be used to infer the strength and direction of selection and to reveal the levels of purifying selection. The ratios of Ka/Ks for all the PCGs are much lower than 1, indicating that all of them are evolving under purifying selection. The genecox1 has a sufficiently large size (1 534 bp) and a low evolutionary rate (0.065) and sequence diversity (Pi=0.175), suggesting that it is under the strongest level of purifying selection.
3.6 Phylogenetic analyses
Phylogenetic trees support the monophyly of the entire Cicadomorpha and each superfamily within Cicadomorpha. It confirms that Membracoidea is a sister group to (Cercopoidea+Cicadoidea) [Bootstrap support values (BS)=100, Bayesian posterior probability (PP)=1.00]. Within Cicadoidea,Tettigarctacrinitaof Tettigarctidae is located at the base of the branch and constituted a sister group to all other cicada species. The monophyly of Cicadidae is strongly supported in all analyses (BS=100, PP=1.00). The inferred relationships in Cicadidae are as follows: Deroterttiginae+(Tibicininae+(Cicadettinae+Cicadinae)) (BS=100, PP=1.00) (Fig. 4: A, B) (no mitogenomic data of Tettigomyiinae are available currently).

Fig. 4 The phylogenetic relationships inferred based on the PCG datasets from 71 Auchenorrhynchan insect mitogenomesA: Phylogenetic relationships inferred based on maximum likelihood and Bayesian inference analyses; B: Phylogenetic relationships among Tettigades spp. (except for T. dumfriesi), which are slightly different from topologies revealed in Fig. A; C: Phylogenetic relationships of taxa at the tribe level within Cicadinae. The numbers at the nodes are the bootstrap (BS) values and Bayesian probability (PP).
In regard to Cicadinae, 19 species are included in the present study, representing nine monophyletic groups (tribes) with well support (BS>75, PP>0.987) (Fig. 4: C). The relationships of these tribes are inferred as: Cryptotympanini+(Fidicinini+(Platypleurini+((Psithyristriini+(Sinosenini+Dundubiini))+(Leptopsaltriini+(Polyneurini+Cicadini))))). The results are generally consistent with previous phylogenetic analyses based on molecular data and/or morphological evidence. The ML and BI trees support that Sinosenini, represented byKarenia, is relatively close toMeimunaof Dundubiini (BS=100, PP=1.00). In addition, the monophyly ofYezoterpnosiain the tribe Leptosaltriini is rejected (BS=100, PP=1.00) (Fig. 4: A, B).
4 DISCUSSION
4.1 Characteristics of mitogenomes of Cicadoidea
The length of mitogenome ofK.caelatata(14 960 bp) is within the range of previously sequenced mitogenomes of Cicadoidea, which range from 13 672 bp (Tettigadesulnaria) to 15 426 bp (Pomponialinearis). Length variation of mitogenomes of Cicadoidea occurs mainly due to variation in the length of the non-coding regions (especially the control region). The gene organization ofK.caelatatamitogenome is consistent with other mitogenomes of Cicadoidea as well as that of the putative ancestral arrangement of insects (Clary and Wolstenholme, 1985). The negative AT-skew and GC-skew on the whole mitogenome ofK.caelatataindicate that T and C are more abundant than A and G (Wangetal., 2015), as is common within Cicadoidea. The spacer oftrnS2-nad1 is common in Cicadoidea, which can also be found in Neuroptera (Jiangetal., 2017), Megaloptera (Beckenbach and Stewart, 2009) and Lepidoptera (Zhangetal., 2019), assumed to contain the binding site for the bidirectional transcription termination factor (DmTTF) (Robertietal., 2003). More research is required to determine the functions and structure of conserved non-coding regions present interspersed between genes in the coding region and the control region among insect mitogenomes.
The incomplete stop codon of PCGs, a single T- or TA, is common in insect mitogenomes. This is presumably completed via post-transcriptional polyadenylation during the mRNA maturation process (Ojalaetal., 1981). The amino acid codons ended with A/T are more frequent than those terminated with G/C, reflecting that a high A+T content occurred. These codon compositions can explain why the entire mitogenomes of insects have a high level of A+T content (Duetal., 2017a, 2017b; Huetal., 2019; Wangetal., 2019). Unlike PCGs with functional annotation features such as possessing start and stop codons, it is difficult to determine the boundaries of rRNA genes from their sequences alone. Therefore, the boundaries of the two rRNA genes in the mitogenome ofK.caelatatawere identified by the boundaries of adjacent tRNA genes, where have neither overlap nor gap between them (Fig. 1). The location, length, the A+T content and the skew of two rRNA genes are much conserved among the mitogenomes of Cicadoidea.
The mismatches in tRNA arms are common in insect mitogenomes, which could be restored during post-transcriptional editing processes (Lavrovetal., 2000). The secondary structure oftrnS1 lacks a dihydrouridine (DHU) arm, which forms a simple loop (Fig. 3). This is a common phenomenon and appeared very early in insect mitogenomes (Wolstenholme, 1992b). The missing of the DHU arm intrnS1 can be functional in the structural compensation mechanism between other arms (Steinberg and Cedergren, 1994). The predicted secondary structures of 22 tRNAs ofK.caelatatamitogenome show that all the arms of tRNAs are generally more conserved than the corresponding loop. In addition, there is an extra unpaired adenine in the anticodon arm oftrnS1 inK.caelatata. This is a common molecular feature of mitogenomes in Cicadoidea, which can be found in the anticodon arm oftrnS1 in many species of Auchenorrhyncha and other suborders of Hemiptera except for Coleorrhyncha (mitogenomes of only six species have been sequenced in Coleorrhyncha) (Huaetal., 2008; Song and Liang, 2009; Liangetal., 2016; Songetal., 2016, 2017; Duetal., 2017a; Su and Liang, 2018, 2019; Suetal., 2018; Huetal., 2019; Xuetal., 2019). This indicates that an unpaired adenine in the anticodon arm oftrnS1 is common in the mitogenomes of hemipteran insects.
The genecox1 has a large size, a low evolutionary rate and a low sequence diversity in Cicadoidea, suggesting that it is under the strongest level of purifying selection. The results of nucleotide diversity analysis in Cicadoidea indicate thatatp8,nad6 andnad2 may be more useful thancox1,cox3 andnad1 for analyzing intraspecies relationships, since they are under a relaxed level of selection when compared with the latter ones.
4.2 Phylogenetic relationships of taxa in Cicadoidea based on mitogenomes
The topological discordances in different phylogenetic trees constructed in the present study should be due to the sample bias. For example, only one species ofMuda, one species ofCicadettanabut 12 species ofTettigadeswere applied to the phylogenetic analyses. Increasing the number of taxon sampling will improve the resolution of the phylogenetic relationships and provide a more comprehensive phylogeny within Cicadoidea when more mitogenomes are available. All the trees constructed in our present study argue removing Cicadatrini (represented byMoganniaandVagitanus) from Cicadinae to Cicadettinae. This has been discussed by Chenetal. (2012) and Wangetal. (2017) based on analysis of morphological characteristics, and Marshalletal. (2018) based on analysis of morphological characteristics and molecular data. Species of Cicadatrini possess timbal covers, an attribute traditionally associated with Cicadinae, but they are closer to the members of Cicadettinae in terms of body size (i.e., small cicadas). Some previous studies on Cicadatrini,e.g., Chenetal. (2012) based on external morphological characteristics, Marshalletal. (2018) based on morphological characteristics and molecular data, andukasik (2019) based on mitogenomic data, also indicated that related members of this tribe are allied to the taxa of Cicadettinae instead of Cicadinae. Furthermore, the monophyly ofYezoterpnosiais rejected in our present study, which is consistent with the results of Leeetal.(2012) and Hilletal. (2021). Therefore, the phylogenetic relationships among taxa ofYezoterpnosiaand other related taxa in Leptosaltriini need to be further investigated in the future.
Regarding the monogeneric tribe Sinosenini, its taxonomic status and the phylogenetic relationship with other taxa have been controversial for a long time. Moulds (2005) placed Sinosenini in Cicadettinae, but no taxa of Sinosenini were included in his phylogenetic analyses. It is the same that no Sinosenini taxa were included in the phylogenetic analyses ofukasiketal. (2019) and Simonetal. (2019). Marshalletal. (2018) conducted phylogenetic analysis of Cicadoidea based on morphological characteristics and molecular data of five genes (i.e.,cox1,cox2,ARD1,EF-1αand 18S rRNA), indicating thatKareniais a member of Cicadinae, but the exact placement was poorly supported. The results of our study based on mitogenomic data indicate thatKareniais closely related toMeimunaof Dundubiini in Cicadinae. Recently, Hilletal. (2021) reconstructed molecular phylogenetic trees of partial Asian cicada tribes using the mitochondrial genecox1 combined with nuclear genesEF-1αandARD1, which revealed thatKareniais most closely related toMacrosemiaof Dundubiini and Sinosenini synonymous with Dundubiini. Although the relationships amongKarenia,MeimunaandMacrosemiaawait further clarification when mitogenomic data ofMacrosemiabecome available, the results of above analyses all show thatKareniais closely related with members of Dundubiini, and thus should be a member of Cicadinae instead of Cicadettinae and merged with Dundubiini. This supports the views of Zhongetal. (2013) based on a comparative study of the salivary glands of cicadas, Lietal. (2015) based on the comparative morphology of the distal segments of Malpighian tubules of cicadas, and Cui and Wei (2018) based on the comparative morphology and ultrastructure of spermatozoa of cicadas.
Concerning other cicadas that possess no timbal organs,ukasiketal. (2019) and Simonetal. (2019) indicated thatPlatypediabelongs to Tibicininae. No other studies have been conducted for the remaining taxa that have no timbals. Combined with results of previous studies, the results of our present study argue that the five genera lacking timbals are not a monophyletic group,i.e., they lost timbal organs independently in the process of evolution (Boulard, 1988; Chenetal., 2012). The feature that timbal organs are completely covered by the timbal covers should not be taken as a synapomorphy of the Cicadinae. Therefore, the higher classification of the Cicadoidea primarily based on structure characteristics associated with sound-producing organs is unreasonable. More phylogenetic analyses should be conducted to confirm the phylogenetic status ofKareniaand other taxa which lack timbals, and the results may improve our understanding of the evolution of sound-producing mechanisms in cicadas.
