
利用Red/ET重组技术构建表达非洲猪瘟病毒CP530R和F317L基因的重组伪狂犬病毒
2021-11-02 11:17邵洋洋乔延召赵琪琪李鸿鑫张新珩蔺文成陈伟国谢青梅华南农业大学动物科学学院广东广州510642
中国兽医学报 2021年9期
孔 洁,邵洋洋,乔延召,赵琪琪,李鸿鑫,张新珩,蔺文成,陈伟国,谢青梅 (华南农业大学 动物科学学院,广东 广州 510642)
自2018年8月以来,我国已有多个省份发生非洲猪瘟(African swine fever,ASF)疫情,ASF给我国养殖行业带来了巨大的挑战。ASF是由非洲猪瘟病毒(African swine fever virus,ASFV)感染引起的猪的一种急性、烈性传染性疾病[1],鉴于目前该疫病在全球多个国家蔓延,开展疫情防控工作、研发新型ASF疫苗已迫在眉睫。ASFV属于双股线型DNA病毒,具有多层结构和二十面体形态,其基因组编码超过150多种病毒蛋白[2-3]。pp62多聚蛋白是ASFV非常重要的一类结构蛋白,该蛋白可以加工合成两个核心蛋白——p35和p15,存在于病毒颗粒的核壳,由CP530R基因编码合成[4]。研究发现,pp220是病毒内部核心壳的主要成分之一,两种多聚蛋白前期的加工都伴随着病毒的组装,当pp62多聚蛋白的表达受到阻遏,间接影响pp220多聚蛋白的离域化[5],引起病毒工厂中具有不同核心发育水平的异常病毒颗粒积聚。未知蛋白(hypothetical proteins)是指缺少功能注释信息的蛋白[6]。在成熟的ASFV粒子中,目前已检测到68种蛋白质,结构蛋白主要参与病毒的复制和病毒粒子的形成,至今仍未知其功能的蛋白质占30%左右,其中包括F317 L[7]。
伪狂犬病是由伪狂犬病病毒(pseudorabies virus,PRV)引起的一种急性传染病,临床上能够引起猪的繁殖障碍。该病毒基因组庞大,全长约145 kb,由于病毒感染宿主范围广,且含有多个非必需基因,可供多种外源基因插入并稳定表达[8]。已有研究表明,复制非必需区基因(TK、PK、gE、gI和gG等)能够作为外源基因的插入位点,缺失突变后可使野生株PRV侵入中枢神经系统[9],显著降低病毒毒力。迄今为止,已有多种标记基因和致病微生物的中和抗原基因在该病毒中获得稳定表达[10],目前该病毒已成为多种新型活载体疫苗的理想载体。
重组病毒活载体疫苗是利用DNA重组等一系列基因工程技术,通过对特定病毒的基因组进行改造,将外源性抗原基因插入到载体病毒中进行表达的一类新型疫苗[11]。“Red/ET重组”是近年发展起来的一种基于同源重组原理在大肠杆菌中直接修饰各类DNA分子的新技术[12]。Red/ET重组技术是利用重组酶Redα/β或RecE/T介导的同源重组对DNA精确修饰的技术[13]。与传统的方法比较,该技术具有不受酶切位点限制,不受基因大小限制、无痕修饰、快速操作等优点。利用该技术可以对多株重组病毒目的基因进行修饰[14-15]。为了减少背景菌落影响,提高筛选效率,利用反向筛选标记ccdB以获得修饰的目的DNA,充分展示了该技术结合反向筛选标记ccdB的可操作性和实行性。本研究利用Red/ET重组工程技术和ccdB反向筛选标记,将ASFV CP530R和F317L同时插入到PRV TK位点,构建表达ASFV CP530R和F317L基因的重组PRV,为ASFV新型活载体疫苗的研制奠定了基础。
1 材料与方法
1.1 主要材料真核表达载体pVAX1购自Invitrogen公司;E.coliDH5α 感受态细胞购自上海唯地生物技术有限公司;Gel Extractin Kit、Plasmid Kit、Cycle Pure Kit均购自OMEGA;限制性内切酶购自Biolabs公司;克隆载体pMD-19T Vector、DNA 聚合酶、DNA Marker均购自TaKaRa公司;PUC57-CMV-CP530R-F317L-BGH质粒由上海生工生物公司合成;质粒pBeloBAC11-cm和表达重组蛋白RecE/RecT的E.coliGBred/GBredA462感受态由山东大学张友明教授惠赠;PRV(Bartha-K61)毒株、rBartha-K61-EGFP(TK)重组毒株、pBeloBAC11-cm-Bartha-K61质粒、Vero细胞、PK-15细胞均由华南农业大学家禽研究室保存备用。
1.2 PCR引物设计及合成以PBR322-Amp-ccdB质粒为模板,扩增含HA同源臂的(TK)-Amp-ccdB-(TK)载体(筛选标记基因片段)。F1:5′-CTCGGCGGGGCGCTGTACGTGCCCGAGCCG-ATGGCGTACTTTAATTAATTTGTTTATTT-TTCTAAATA-3′;R1:5′-GGAAGCACACCGTC-GCGGCCACCGGGTGGCGGTCAAAGACTTAA-TTAATTATATTCCCCAGAACATCAG-3′(限制性内切酶PacⅠ酶切位点);以PUC57-CMV-CP530R-F317L-BGH质粒为模板,扩增含HA同源臂的基因片段((TK)-CMV-CP530R-F317L-BGH-(TK))。F2:5′-CTCGGCGGGGCGCTGTACGTGCCCGAGCCGATGGCGTACTTTAATTAAGA-CATTGATTATTGACTAGT-3′;R2:5′-GGAAGCACACCGTCGCGGCCACCGGGTGGCGGTCA-AAGACTTAATTAATGTCCATAAAACCGCC-CAGT-3′(限制性内切酶Pac Ⅰ酶切位点)。
PCR反应体系:2×PrimeSTAR Max DNA Polymerase 25 μL,Template DNA 1 μL,引物(F/R)各2 μL,ddH2O补足50 μL。PCR扩增程序:98℃ 2 min;98℃ 10 s,55℃ 5 s,72℃ 30 s,35 个循环;72℃ 3 min,PCR产物进行1%琼脂糖凝胶电泳,观察结果并测序。
1.3 重组病毒的构建及两步法转化由上海生工生物合成的ASFV抗原基因来源于中国流行毒株Pig/CN/HLJ/2018(GenBank:MK333180.1),命名为:PUC57-CMV-CP530R-F317L-BGH。以PBR-322-Amp-ccdB为模板,(TK)Amp-ccdB-F/(TK)Amp-ccdB-R为引物,PCR扩增(TK)-Amp-ccdB-(TK)筛选标记基因片段。以基因合成的质粒为模板,利用通用引物(TK)CMV-F/(TK)BGH-R,PCR方法扩增外源基因片段,命名为:(TK)-CMV-CP530R-F317L-BGH-(TK)。
将制备好的基因片段(TK)-Amp-ccdB-(TK)与pBeloBAC11-cm-Bartha-k61载体进行同源重组,将RecE/RecT重组蛋白转化至感受态GB redA462,涂布于Amp抗性的LB平板,重组质粒命名为 pBeloBAC11-cm-Bartha-k61-(TK)-Amp-ccdB-(TK),完成与环状载体第1步重组病毒的转化。将制备好的基因片段(TK)-CMV-CP530R-F317L-BGH-(TK)与pBeloBAC11-cm-Barthak61-(TK)-Amp-ccdB-(TK)质粒进行同源重组,将RecE/RecT重组蛋白转化至感受态GB red,涂布于氯霉素抗性的LB平板,进行第2步重组病毒的转化。
1.4 感染性克隆的筛选与鉴定电转完成后立即加入1 mL SOC培养基至电击杯中,将菌体重悬并转移至2 mL离心管中,37℃,260 r/min培养1 h完成重组及抗性复苏。3 000 r/min离心1 min沉淀菌体,保留部分上清并吹打重悬,涂布含有Amp抗性的LB平板,37℃培养12~16 h。挑取单克隆菌落,37℃,260 r/min培养12 h。以(TK)CMV-F/(TK)BGH引物进行菌液PCR初步鉴定,提取质粒进行Pac Ⅰ酶切鉴定,鉴定正确的质粒送至生工生物工程(上海)股份有限公司进行测序鉴定,鉴定正确的重组质粒命名为:pBeloBAC11-cm-Bartha-k61-(TK)- CP530R-F317L-(TK)。
1.5 病毒拯救鉴定用鉴定正确的重组质粒pBeloBAC11-Bartha-K61-(TK)CMV-CP530R-F317L-BGH按照LipoTM3000说明书转染Vero细胞,37℃,5% CO2培养48 h,收获细胞及上清,接种至PK-15细胞,盲传5代,收获病毒液进行拯救病毒鉴定。病毒感染后出现空泡等典型噬斑状态,酶切正确的克隆命名为:rBartha-K61-CP530R-F317L,抽提病毒基因组DNA,PCR扩增基因片段并进行鉴定。
取rBartha-K61-CP530R-F317L株病毒液接种至PK-15细胞(MOI=1),收取细胞,一抗使用鼠源Flag标签蛋白多克隆抗体(1∶750稀释)进行孵育,二抗使用FITC标记的山羊抗小鼠IgG(1∶10 000稀释)进行孵育,Western blot鉴定蛋白表达。
1.6 感染性克隆的稳定性试验将感染性克隆rBartha-K61-CP530R-F317L以MOI=1接种PK-15细胞,48 h产生明显病变,反复冻融3次后收获病毒,一步生长曲线测定病毒TCID50。连续传代20次,每隔5代提取感染性克隆的基因组DNA,按照1.4方法进行PCR鉴定,第20代按照1.5方法进行Western blot鉴定。
2 结果
2.1 PCR扩增及测序含有同源臂的目的基因扩增结果显示,目的条带大小约为2 671 bp(图1A),扩增片段与预期大小一致,胶回收目的条带并进行克隆测序;Amp-ccdB筛选标记基因扩增结果显示,目的条带大小约为1 307 bp(图1B),扩增片段与预期大小一致,胶回收目的条带并进行克隆测序。
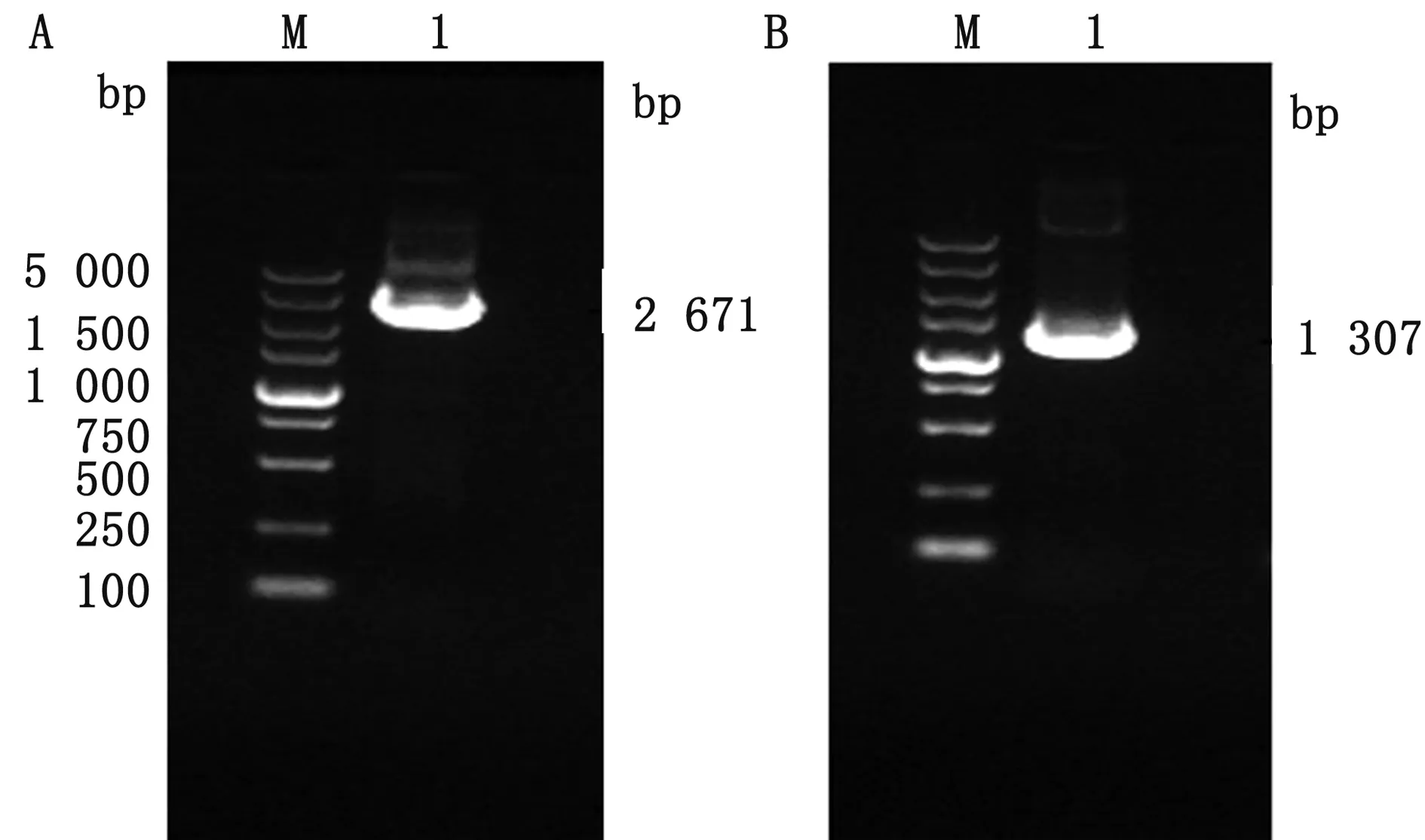
A.含有同源臂的目的基因;B.Amp-ccdB筛选标记基因图1 含有同源臂的CP530R-F317L基因和Amp-ccdB片段扩增结果
2.2 真核表达重组质粒鉴定将目的基因定向克隆到真核表达载体上,使用质粒抽提试剂盒对阳性菌液进行抽提,限制性内切酶PacⅠ对抽提质粒进行酶切鉴定,结果显示在3 646,7 516,58 803,78 782 bp处可见4条与预期大小一致的条带(图2),胶回收目的条带并进行测序验证,测序结果显示正确,说明重组质粒构建成功。

图2 真核表达重组质粒的酶切鉴定
2.3 真核表达重组质粒筛选将鉴定正确的重组质粒转染至Vero细胞后48 h收获细胞及上清,接种PK-15细胞,盲传至第5代时,PK-15细胞膨大变圆,随之细胞变性圆缩,最后细胞脱落聚集,形成空斑等典型CPE现象(图3),将该拯救毒株命名为rBartha-K61-CP530R-F317L,连续传代20代后未发现变异。
A.空白对照细胞;B.感染后的细胞病变图3 重组病毒质粒感染PK-15细胞病变
2.4 感染性克隆的稳定性试验
2.4.1感染性克隆的蛋白鉴定 以rBartha-K61-CP530R-F317L为模板,PCR扩增含同源臂的目的基因,目的条带与预期大小一致。为验证感染性克隆的蛋白表达情况,将rBartha-K61-CP530R-F317L感染性克隆,以MOI=1接种至细胞培养6孔板单层PK-15细胞,使用带有HA标签多克隆抗体进行Western blot检测,结果显示(图4),rBartha-K61-CP530R-F317L感染性克隆组检测到HA标签蛋白,疫苗株Bartha-K61未检测到HA标签蛋白。

图4 感染性克隆的蛋白鉴定
2.4.2感染性克隆的一步生长曲线 利用Reed-Muench法[16]计算Bartha-K61疫苗株、rBartha-K61-EGFP(TK)、rBartha-K61-CP530R-F317L重组病毒不同时间点的TCID50,绘制一步生长曲线(图5)。重组病毒能在PK-15易感细胞上快速增殖,病毒滴度峰值可达105.8TCID50/mL,与原始毒株Bartha-K61存在差异,但与rBartha-K61-EGFP(TK)重组毒株无明显差异(表1)。
图5 感染性克隆的一步生长曲线

表1 不同时间点病毒滴度测定(lgTCID50)
3 讨论
自2018年8月至今,ASF在全球范围内的广泛传播造成了巨大的经济损失,ASF疫情的流行特征及动态变化迫切要求有效的预防控制措施,开发及研制高效、安全的新型疫苗具有重要的研究意义。在宿主和病毒长期共存的漫长过程中,病毒进化了各种免疫逃逸机制以逃避宿主的免疫应答[17-19],ASFV拥有复杂且庞大的基因组,编码多种免疫逃逸蛋白抑制宿主免疫应答,使得该病尚未有有效疫苗问世[20-22],现阶段在做好ASFV防控保障工作的同时,越来越多的国家将工作重心趋向于研究新型ASF疫苗以此进行免疫评估。
目前,针对ASF预防在畜禽研究中的范围和内容十分广泛,尤其是在临床病例中不同类型疫苗的开发及应用。早期ASF疫苗研制的重心主要聚焦于灭活疫苗,但前期研究表明,ASFV灭活疫苗虽可刺激机体产生不同强度的免疫应答,但仍无法诱导宿主产生中和性抗体,尚未呈现良好的中和作用。BLOME等[23]研究发现,使用新型佐剂(PolygenTM或Emulsigen®-D)与灭活的ASFV疫苗配伍,均可检测到ASFV特异性抗体,但未观察到免疫的保护作用。与基因缺失减毒活疫苗相比,DNA疫苗是将病毒编码的主要蛋白基因通过克隆等一系列方式插入真核表达载体,构成重组真核表达载体刺激机体产生体液免疫和细胞免疫应答。ARGILAGUET等[24]将ASFV血凝素(sHA)的胞外域与两种病毒抗原——p54和p30融合后,虽可刺激机体产生体液免疫和细胞免疫,但无法起到攻毒保护,研究者进一步将新的重组质粒pCMV-UbsHAPQ与泛素融合的病毒决定簇——sHA,p54和p30,发现仅能够改善并刺激机体产生部分免疫保护。安全性是影响弱毒活疫苗临床应用最关键的影响因素之一[25],ASFV弱毒活疫苗主要分为3类——传代致弱、重组致弱和天然致弱。KRUG等[26]将ASFV-G野毒株在Vero细胞中连续传代110次,体外评估获得病毒在Vero细胞和猪原代巨噬细胞中复制的能力,体内评估对猪的毒性,病毒全长序列分析显示出病毒具有明显的缺失。重组活载体疫苗是指将病毒的保护性抗原通过分子生物学技术插入到病毒载体上,以期获得有效的免疫反应和免疫保护。引起体液和细胞介导免疫反应的策略是使用病毒载体。LOKHANDWALA等[27]首次采用“鸡尾酒疗法”,将多种抗原混合物与佐剂配伍使用,快速诱导了ASFV抗原特异性抗体及针对所有抗原的细胞免疫反应,该项研究首次证明使用Ad-ASFV多种抗原共同诱导商业猪抗原特异性CTL反应。现阶段,仍需要进行大量的临床试验验证重组活载体疫苗的安全性和必要性。
Red/ET重组技术是建立在同源重组基础上,对大肠杆菌中DNA分子进行高效修饰的遗传工程技术。Red/ET技术由重组酶——Redα/β或RecE/T催化DNA分子,Redα/β是由来源于λ噬菌体的pL操纵子的redα和redβ基因编码的重组蛋白;RecE/T是分别由来源于E.coliRac前噬菌体的recE和recT基因编码的重组蛋白[28]。研究发现,针对“线线”重组,重组酶RecE/T比Redα/β介导效率更高,而 Redα/β介导“线环”重组效率更高,Redα/β蛋白能够高效介导线性DNA分子和环状DNA分子之间发生同源重组[29-30]。利用Red/ET重组对目的基因进行修饰,为了减少背景菌落影响,提高筛选效率,科研人员提出了“两步走”的策略[31]。常用的正筛选标记包括各种抗生素抗性基因等,负筛选标记包括sacB、rspL、galK和ccdB等[32]。ccdB会通过促进遗传物质的广泛损伤来快速杀灭细菌,作用于的靶标是解旋酶的GyrA亚基,阻止DNA复制和转录,从而导致细胞死亡。
本研究利用Red/ET重组工程技术和ccdB反向筛选技术,将ASFV特定基因——CP530R-F317L,定向插入到PRV TK位点,最终表达ASF的特定抗原基因,重组活载体疫苗具有良好的遗传稳定性和增殖型,为新型ASFV疫苗的开发提供了理论基础。
猜你喜欢
传染病信息(2022年6期)2023-01-12
环球时报(2022-09-20)2022-09-20
心电与循环(2021年4期)2021-08-03
今日农业(2020年24期)2020-12-15
食品科学(2018年10期)2018-05-23
兽医导刊(2016年12期)2016-05-17
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年12期)2015-06-10
中国当代医药(2015年9期)2015-03-01
现代检验医学杂志(2015年4期)2015-02-06
