
抗艾滋病药物洛匹那韦的合成及结构表征①
2021-11-02 14:29孙远东高永好杨士伟司友琳
佳木斯大学学报(自然科学版) 2021年5期
孙远东, 何 勇, 高永好, 杨士伟,*, 司友琳
(1.蚌埠医学院,安徽 蚌埠233030;2.合肥华方医药科技有限公司,安徽 合肥230088)
0 引 言
艾滋病是由人类免疫缺陷病毒感染引起的全身性免疫功能丧失综合征[1-2]。HIV蛋白酶作为潜在的抗艾滋病病毒药物设计的靶向目标,开发更加高效更加成熟的HIV蛋白酶抑制剂(PI)药物是目前抗艾滋病药物研发的热门方向[3-5]。目前,市场上诞生的HIV蛋白酶抑制剂(PI)药物包括:洛匹那韦、沙奎那韦、利托那韦、茚地那韦、安普那韦、奈非那韦等品种[6-9]。其中,雅培公司于2000年申请上市了洛匹那韦和利托那韦复方制剂品种,利用洛匹那韦对突变型和野生型HIV-1PR高活性,在一定程度上解决了利托那韦的耐药性问题,从而达到阻断HIV复制的目的[10-12]。通过酰胺化反应和缩合反应合成制备纯度较高的洛匹那韦,83%收率,纯度99.76%,其结构通过核磁共振仪表征。
1 实验部分
1.1 仪器与试剂
(2S,3S,5S)-2一胺基-3一羟基.5.(叔丁氧羰基胺基)-1,6-二苯基己烷(BDH,含量98.9%),麦克林试剂有限公司;(2S)-(1-四氢嘧啶-2-酮)-3-甲基丁酸,2,6-二甲基苯氧乙酸(纯度>98%),麦克林试剂有限公司;三氟乙酸(TFA),安耐吉试剂有限公司;二氯亚砜(SOCl2),安耐吉试剂有限公司;1-羟基苯并三氮唑(HOBT),麦克林试剂有限公司;1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC盐酸盐),安耐吉试剂有限公司;N,N-二甲基甲酰胺(DMF),安耐吉试剂有限公司;乙酸乙酯(Ethyl Acetate)、二氯甲烷(DCM)、四氢呋喃(THF)、无水硫酸钠(Na2SO4)、碳酸氢钠(Na HCO3)、氢氧化钠(NaOH)、氢氧化钾(KOH)、氢氧化锂(LiOH),AR,国药集团化学试剂有限公司。
AB4000三重四级杆串联质谱仪(ESI+源)、傅里叶红外光谱仪Nicolet 6700,赛默飞世尔科技(中国)有限公司;AVⅢ400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标,瑞士Bruker公司。
1.2 实验方法
1.2.1 洛匹那韦利中间体(I)的合成
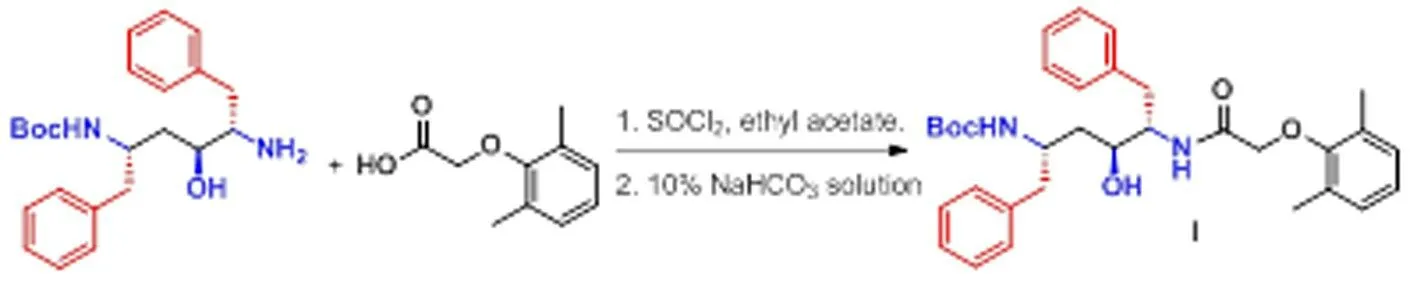
图1 洛匹那韦中间体(I)的合成
250ml三口瓶中加入14.2g(78.8 mmol,纯度>98%)2,6-二甲基苯氧乙酸,50 m L乙酸乙酯,室温搅拌溶解,室温下自恒压滴液漏斗滴加12.2g(102 mmol)SOCl2和3滴DMF溶液(15 min滴完),升温至50℃反应4h。然后减压蒸出溶剂乙酸乙酯后,加入60ml乙酸乙酯溶解待用。1L三口瓶中加人26.2g(68 mmol)的化合物(2S,3S,5S)-2一胺基-3一羟基.5.(叔丁氧羰基胺基)-1,6-二苯基己烷(BDH),300ml乙酸乙酯,300ml水和31g碳酸氢钠固体,室温搅拌。待固体溶解后,剧烈搅拌向该两相溶液中滴加上述酰氯的乙酸乙酯溶液(15 min滴完),于室温下反应5 h,HPLC监控原料BDH反应完全后。分液,有机相加人10%碳酸氢钠水溶液200ml,于室温搅拌30 min后分液除去水相,再用200ml水洗,有机相加入40g无水硫酸钠,干燥,过滤,浓缩,真空干燥后得到白色固体30g,收率:80%,熔点:170.6℃~171.0℃。
1.2.2 洛匹那韦利中间体(II)的合成

图2 洛匹那韦中间体(II)的合成
氮气保护下,10.0 g(18.3 mmo1)中间体I、100m L二氯甲烷溶解后,于0~5℃下向该溶液中滴加40ml二氯甲烷溶解的20.8 g(183 mmo1)三氟乙酸(约40 min滴完),此过程需缓慢滴加,预防副反应的发生。滴加完后升至20-25℃反应4 h,HPLC监控原料中间体1反应完全。减压蒸馏回收未反应完全的三氟乙酸,蒸干后加入二氯甲烷200 ml溶解,10% Na HCO3水溶液200 ml洗涤至p H=8-9,然后分液。对有机相再以200 ml水洗一次,无水硫酸钠干燥,过滤,浓缩,得到淡黄色油状物,加入到乙酸乙酯/正庚烷(2:3)混合溶剂30 ml,降温至0℃,搅拌析晶,过滤,40℃真空干燥,得到7.1g白色固体,熔点:131.6-132.6℃。
1.2.3 洛匹那韦的合成
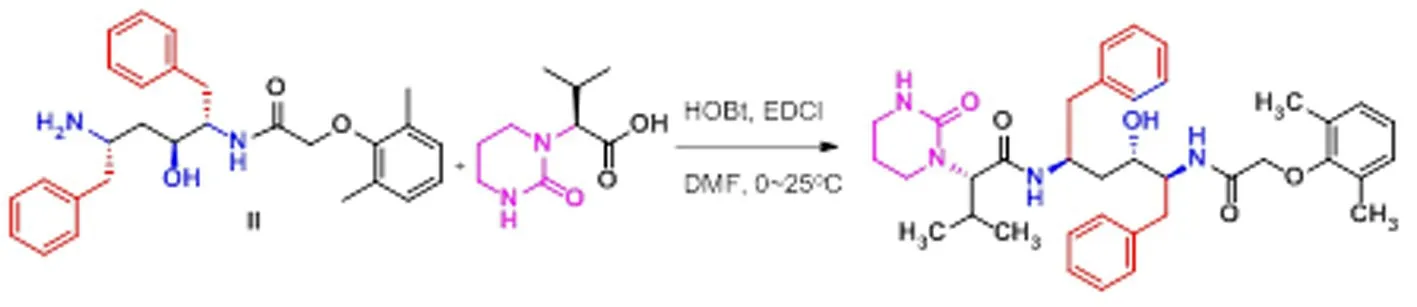
图3 洛匹那韦的合成
氮气保护下,40mL干燥DMF、1.1g(10.9mmol)干燥三乙胺,4.2g(9.1 mmo1)中间体II,2.0g(10 mmol])(2S)-(1-四氢嘧啶-2-酮)-3-甲基丁酸,1.66g(12.3 mmo1)1-羟基苯并三氮唑(HOBT)后,于0℃下搅拌20 min后向此混合,分批加入2.1g(10.9 mmo1)1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC盐酸盐),升至20-25℃反应过夜(不低于12 h),HPLC监控中间体2反应完全。减压蒸出DMF后加入100 ml乙酸乙酯、100 ml 1.0 mol/l稀盐酸洗涤,分液,对有机相依次以150 m L 5%(wt%)Na HCO3,150 ml蒸馏水洗涤,无水硫酸钠干燥和活性炭脱色、过滤、浓缩,得淡黄色到油状物,用乙酸乙酯和正己烷30g(乙酸乙酯∶正己烷=1∶1)结晶,抽滤,40℃烘干,得到白色固体,收率83%,HPLC纯度97.76%。:1H NMR(400 MHz,DMSO-d6)δ7.55(d,J=8.8 Hz,1H),7.46(d,J=8.8 Hz,1H),7.25(m,4H),7.19(m,3 H),7.12(m,3H),7.01(m,2H),6.93(m,1H),6.30(m,1H),5.00(d,J=5.7 Hz,1H),4.32(d,J=11.2 Hz,2H),4.26-4.16(brs,1H),3.35(s,1H),3.04(t,J=6.0 Hz,2 H),2.92(t,J=6.0 Hz,2H),2.81(m,2H),2.66(m,1H),2.57(m,1 H),2.15(s,6 H),2.01(m,1H),1.52(m,4H),0.85(t,J=6.6 Hz,2H),0.76(dd,J=9.6,6.5 Hz,6 H);13CNMR(101 MHz,DMSO)δ169.29,167.28,155.48,154.50,139.01,138.82,130.20,129.18,129.08,128.76,127.95,127.81,125.85,125.53,124.19,70.23,68.38,61.46,52.57,46.72,37.87,31.21,28.31,25.44,22.04,21.68,19.53,18.65,15.83,13.88.;Mp(DSC):133-135℃.;EIMS:629.4[M+H]。
2 结果与讨论
2.1 洛匹那韦中间体(I)合成反应条件的考察
通过二氯亚砜作酰化试剂对2,6-二甲基苯氧乙酸在无添加剂存在条件下进行酰氯化反应,在二氯甲烷(DCM)溶剂中25℃反应12h,无中间体I产生(表1,序号1)。相同反应温度,添加剂选用N,N-二甲基甲酰胺(DMF),分别在乙酸乙酯溶剂和二氯甲烷溶剂中反应12h,收率分别为16%(表1,序号2)和19%(表1,序号3)。反应温度提高到50℃,在无添加剂存在条件下进行酰氯化反应12h,收率提高到41%(表1,序号4)。选用DMF作为添加剂,乙酸乙酯和二氯甲烷作为溶剂反应12h,收率可以提高到59%(表1,序号5)和66%(表1,序号6),其中二氯甲烷溶剂条件下反应产生杂质较多。反应温度为50℃,二氯甲烷中DMF作为添加剂反应8h,中间体I的分离收率达到80%(表1,序号7)。继续提高温度到75℃,反应收率降低,推测原因是温度越高,反应杂质继续增多(表1,序号8-10)。最终获得最佳反应条件为以二氯甲烷为溶剂,DMF作为添加剂,反应温度为50℃,反应时间为8h,反应收率为80%(表1,序号7)。
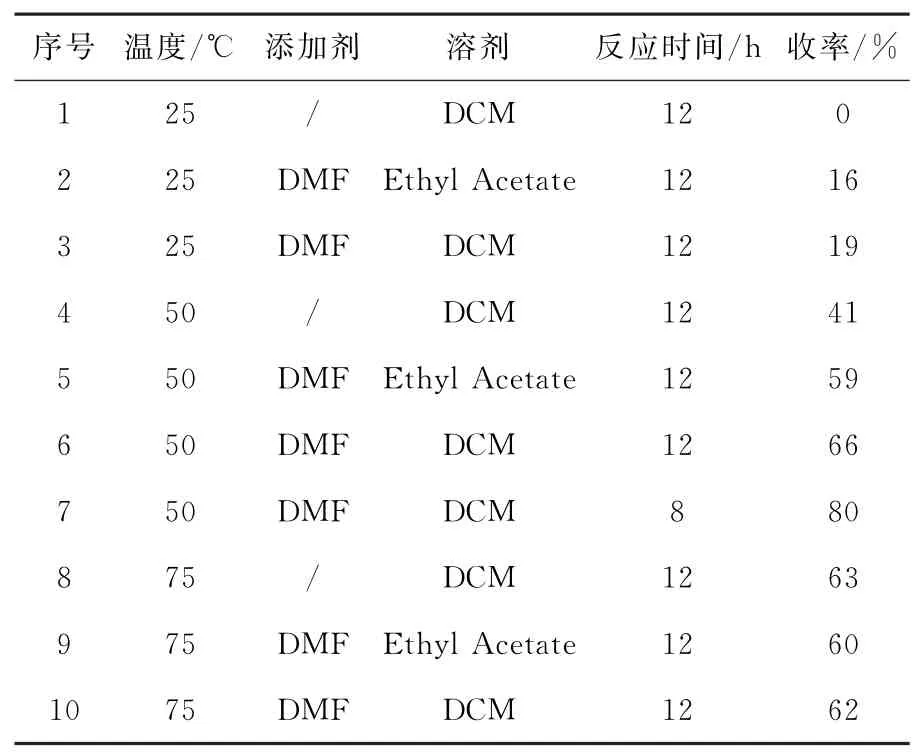
表1 洛匹那韦中间体I合成反应条件的优化
2.2 洛匹那韦的合成反应条件的考察
通过三氟乙酸(TFA)对洛匹那韦中间体I进行脱Boc制备获得的洛匹那韦中间体II和(2S)-(1-四氢嘧啶-2-酮)-3-甲基丁酸在DCC添加剂存在条件下进行酰胺化反应,在二氯甲烷(DCM)溶剂中-5℃反应12h,洛匹那韦分离收率只有9%(表1,序号1)。相同反应温度,反应提高到24h,收率为12%(表1,序号2)。反应温度提高到0℃和反应溶剂更换为乙酸乙酯,反应12h,无洛匹那韦产生(表1,序号3),二氯甲烷作为溶剂反应12h,收率为15%(表1,序号4),添加剂更改为EDCI,反应收率为21%(表1,序号5),说明EDCI对酰胺化反应具有促进作用。反应温度提高为25℃,EDCI作为添加剂,二氯甲烷为反应溶剂,反应24h,洛匹那韦的分离收率提高到29%(表1,序号6),溶剂改为乙酸乙酯反应12h,洛匹那韦的分离收率只有11%(表1,序号7),说明反应溶剂对该反应的影响较大。将反应溶剂更改为DMF,反应温度为25℃,反应时间为12h,18h,24h,洛匹那韦的分离收率分别为46%,59%和83%(表1,序号8-10)。相同反应温度,DMF做溶剂,添加剂从EDCI更改为SOCl2,反映4h,原料未反应完全,洛匹那韦的分离收率为62%(表1,序号11),提高温度到50℃,收率提高到66%(表1,序号12)。反应温度为50℃,EDCI为添加剂,在DMF中反应24h可以制备得到84%收率(表1,序号13),但考虑到原子经济性问题,最终获得最佳反应条件为以DMF为溶剂,EDCI作为添加剂,反应温度为25℃,反应时间为24h,反应收率为83%,HPLC检测产品纯度为99.76%(表1,序号10)。
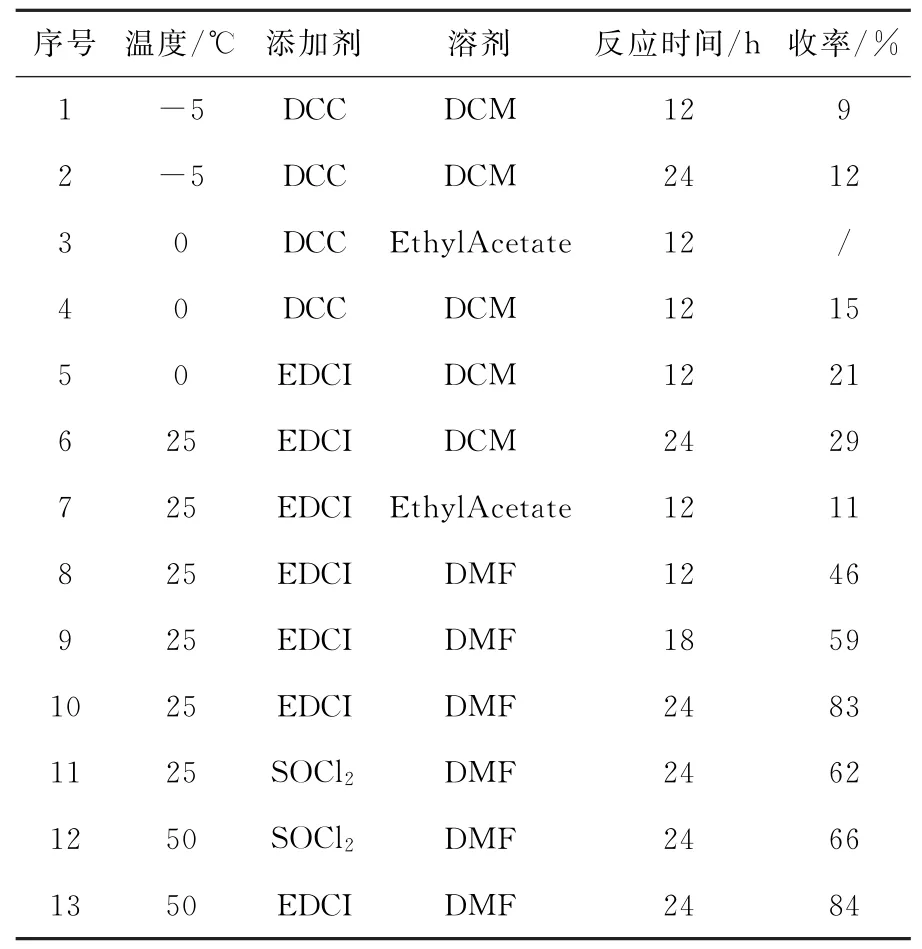
表2 洛匹那韦的合成反应条件优化
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
读与写·下旬刊(2018年6期)2018-07-14
山东工业技术(2018年9期)2018-05-26
科学与财富(2017年17期)2017-06-16
化学教学(2015年12期)2015-12-12
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
股市动态分析(2015年12期)2015-09-10
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
