
热休克蛋白70调控Bcl-2/Bax对缺氧性肺高压新生大鼠肺血管内皮细胞凋亡及肺动脉压力的作用
2021-10-30 10:15李明霞
新疆医科大学学报 2021年9期
赵 倩,曹 静,李明霞
(新疆医科大学1儿科学院,2第五附属医院新生儿科,乌鲁木齐 830011)
新生儿缺氧性肺动脉高压(hypoxia pulmonary hypertension,HPH)是严重威胁新生儿健康的常见急危重症[1],发病机制不清,早期肺血管痉挛病变可逆,到疾病晚期肺血管重塑为不可逆改变,救治困难[2]。该病的起始因素可能是早期的肺血管内皮细胞凋亡、损伤,而晚期促凋亡和抑凋亡细胞之间平衡失调促进了该病的进一步发展;因此调控细胞凋亡的相关蛋白可能参与了该病的发病机制[3]。热休克蛋白70(heat shock protein 70,HSP70)是广泛存在于真核和原核生物中的一种最敏感的应激性保护蛋白,其通过促进细胞存活、抑制细胞凋亡参与肿瘤等疾病的发病过程[4],本课题组前期研究发现,HPH 新生大鼠肺组织中HSP70 表达升高,降低了肺动脉压力及肺血管重塑[5],提示该因子在HPH 疾病中发挥了重要的保护性作用,然而HSP70 是否通过调控及如何调控肺血管内皮细胞凋亡,从而参与该保护性机制仍不清楚,需进一步验证。另有研究表明Bcl-2 和Bax之间表达失调是HPH 动物模型中肺血管内皮细胞凋亡的重要原因,而HSP70 是否作为二者的上游调控因子参与了这一机制目前不确定[6]。本研究拟在前期研究[5]基础上通过腺病毒转染提高HPH 新生大鼠肺组织中HSP70 表达,并通过KNK437 抑制肺组织中HSP70 表达,探讨其对Bcl-2 和Bax 表达水平的影响及其对HPH 新生大鼠肺血管内皮细胞凋亡和肺动脉压力的影响,为新生儿HPH的治疗寻求新的潜在靶点。
1 材料与方法
1.1 实验动物、试剂及设备
1.1.1 实验动物 128只wistar新生大鼠,体重20~30 g,来自新疆医科大学实验动物中心,实验动物许可证号:SCXK(新)2011-0004。
1.1.2 试剂及设备 试剂anti-Bcl-2 (Abcam ab59348)、anti-Bax (Abcam ab53154 )、anti-HSP70 (Abcamab 2787);纯化的Ad-HSP70-EGFP(Enhanced green flu⁃orescentprotein,EGFP)(增强型绿色荧光蛋白标记的HSP70 腺病毒转染液)、Ad-CON-EGFP(增强型绿色荧光蛋白标记的空腺病毒转染液)及KNK437(上海西格玛奥德里奇贸易有限公司)主要设备有小动物呼吸机( HX-200 型,成都泰盟科技有限公司)、YMS-51 氧浓度控制系统(上海玉研科学仪器有限公司)、透射电子显微镜(JEOL-1230 型,日立,日本)、BP-6八导无创血压监测系统(成都泰盟科技有限公司)、荧光共聚焦显微镜(Leica公司,德国)。
1.2 方法
1.2.1 实验分组及目的病毒转染目标基因 将Wistar新生大鼠128只按随机数字表分为对照组和缺氧组,缺氧组造HPH 模型,按干预方法分为HPH 组、HSP70+HPH 组和KNK437+HSP70+HPH 组(KNK437为HSP70 高效抑制剂)。每组分为3、7、10 、14 d 观察点,每个观察时间点均为8 只新生大鼠。将HSP70+HPH 组和KNK437+HSP70+HPH 组新生大鼠经口气管插管注入Ad-HSP70-EGFP,注射剂量2 uL×108PFU/只,HPH 组和对照组经口气管插管注入同等病毒滴度的Ad- CON-EGFP,每日KNK437+ HSP70+HPH组腹腔注射KNK437,注射剂量30 µg/只。
1.2.2 建立新生大鼠HPH 模型 各组预处理后,向低氧舱内冲入均匀的氮氧混合气,通过通气孔维持低氧舱内外压力平衡,并持续检测低氧舱内氧浓度、湿度和温度,维持氧浓度在(10±0.5)%, 湿度60%~70%,温度22℃~25℃,同时使用钠石灰和无水氯化钙分别吸收二氧化碳和水,低氧舱内母鼠自由饮水与进食。对照组氧浓度为21%,余同HPH组。
1.2.3 测定平均RVSP 按照不同观察时间点将新生大鼠常规麻醉、固定、备皮、消毒、气管插管,行机械通气(小动物呼吸机参数设置为呼吸频率100~110次/min、潮气量4~6 mL/min,监测实验新生大鼠尾部血氧饱和度,使其维持在85%~95%)。作胸骨前U 型切口,打开胸腔,充分暴露心脏,在右心室心尖部用静脉穿刺置管针刺入,针头另一端接压力传感器,通过八导无创血压检测系统记录平均右心室收缩压(RVSP),测压后立即处死大鼠,留取肺组织标本以备后续实验。
1.2.4 荧光显微镜观察各组内携带HSP70 腺病毒的定位部位 各组各观察时间点新生大鼠处死后立即留取3 mm× 3 mm× 3 mm 的肺中叶肺组织,PBS 清洗,4%多聚甲醛液固定4 h,PBS冲洗3~4次,蔗糖溶液浸泡过夜,OTC包埋,冰冻切片机切为厚度约为6 µm 的冰冻切片,荧光显微镜下避光观察腺病毒携带的绿色荧光表达情况。
1.2.5 透射电镜观察肺血管内皮细胞凋亡变化 各组不同观察时间点新生大鼠测定平均RVSP 后,立即处死,迅速留取大小约1 mm× 1 mm× 1 mm 肺组织放入4℃2.5% 戊二醛溶液固定2 h,常规铅轴电子染色、包埋、切片,厚度小于100 µm。使用透射电子显微镜观察新生大鼠肺血管内皮细胞凋亡变化。
1.2.6 免疫组化评价新生大鼠肺组织内HSP70、Bcl-2 和Bax 表达情况 各观察时间点新生大鼠处死后,留取肺组织标本甲醛固定1 w,制作石蜡切片。石蜡切片常规脱蜡至水,热抗原修复。一抗孵育过夜,二抗孵育,二氨基联苯胺( DAB ) 显色,光学显微镜采图。 利用Image-Pro Plus 软件检测平均光密度值(IOD/Area)评价蛋白表达情况。
1.3 统计学处理采用SPSS 26.0 软件包进行数据分析。实验数据计量资料描述以均数±标准差(±s)表示,多组间比较采用单因素方差分析(ANOVA)检验。检验水准α=0.05。
2 结果
2.1 各组新生大鼠平均RVSP 变化HPH 组在缺氧3 d 即可观察到平均RVSP 明显增高,随缺氧时间延长,各观察时间点均高于对照组(P<0.05),提示HPH新生大鼠模型构建成功;缺氧3、7、10 d,HSP70+HPH组平均RVSP 低于HPH 组和KNK437+HSP70+HPH组(P<0.05),与对照组比较差异无统计学意义(P>0.05),缺氧14 d各实验组平均RVSP对比差异无统计学意义(P>0.05),但均高于对照组(P<0.05)。见表1。
表1 各组新生大鼠平均RVSP变化及单因素方差分析结果(±s,mmHg)

表1 各组新生大鼠平均RVSP变化及单因素方差分析结果(±s,mmHg)
注:与对照组比较,aP<0.05;与HPH组比较,bP<0.05;与HSP70+HPH组比较,cP<0.05
组别对照组HPH组HSP70+HPH组KNK437+HSP70+HPH组Fp n 8 8 8 8 3 d 17.54±0.43 21.84±0.66a 17.50±0.41b 21.67±0.73ac 109.38<0.001 7 d 18.35±0.41 24.24±0.39a 18.27±0.36b 23.86±0.24ac 519.59<0.001 10 d 19.80±0.40 26.12±0.37a 20.15±0.28b 25.70±0.72ac 315.90<0.001 14 d 21.37±0.38 28.31±0.54a 27.29±2.17a 28.52±0.42a 51.44<0.001
2.2 携带HSP70 腺病毒在新生大鼠肺组织中的定位及免疫组化评估HSP70 表达情况缺氧3、7、10 d各组绿色荧光随时间延长而逐渐减弱,绿色荧光位于细支气管周围,缺氧14 d 各组均未见明显荧光。见图1。缺氧3、7、10 d HPH 组新生大鼠肺组织中HSP70表达高于同日龄对照组(P<0.05),缺氧14 d差异无统计学意义(P>0.05);缺氧3、7 d HSP70+HPH组HSP70 表达高于HPH 组(P<0.05),缺氧10、14 d 高于HPH组,但差异无统计学意义(P>0.05),各观察时间点均高于对照组(P<0.05);缺氧3、7、14 d KNK437+HSP70+HPH组HS表达均低于HPH组(P<0.05),各观察时间点均低于HSP70+HPH 组(P<0.05),较对照组差异无统计学意义。见图2、表2。

表2 各组新生大鼠肺组织HSP70-IOD/Area及单因素方差分析结果
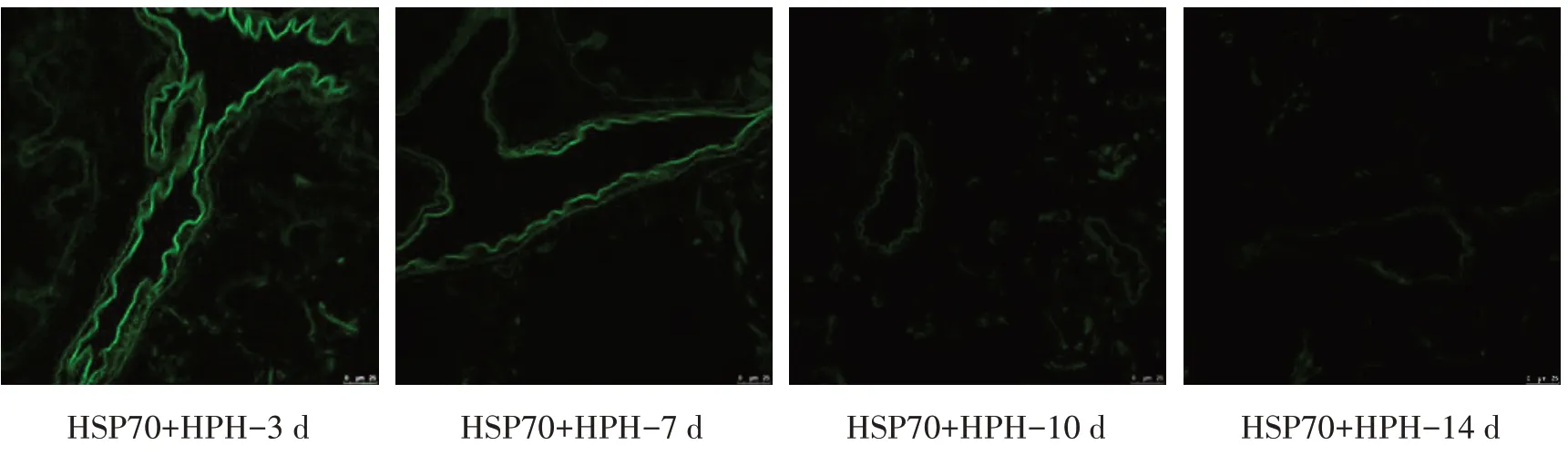
图1 荧光共聚焦显微镜下观察HSP70+HPH组新生大鼠肺组织中的绿色荧光(×200)

图2 各组新生大鼠肺组织中HSP70免疫组化染色(×200)
2.3 透射电镜下观察肺组织内皮细胞凋亡变化对照组肺组织内皮细胞扁平、形状规则,细胞核、染色体形态和分布均正常,未见凋亡改变;HPH 组缺氧3 d 可观察到内皮细胞发生凋亡形态,细胞体积小,质着色深,少数染色质浓缩、边移,缺氧7、10 d 染色质呈境界分明的半月状、块状或完全凝聚附于核模,但缺氧14 d 凋亡减少,以胶原纤维增生为主;HSP70+HPH 组在缺氧3 d 时肺血管内皮细胞的细胞核形态和细胞质分布比较正常,缺氧7、10、14 d 肺血管内皮细胞的细胞质发生变移、凝聚等凋亡和增生变化,其凋亡和增生程度均低于同日龄HPH 组;KNK437+HSP70+HPH组缺氧3 d可观察到染色质浓缩,随着缺氧时间延长,逐渐观察到凋亡减少和增生出现,其程度高于同日龄HSP70+HPH 组,较同日龄HPH 组无明显变化。见图3。

图3 HPH新生大鼠缺氧3d肺血管内皮细胞凋亡变化(×40 000)
2.4 免疫组化评估Bcl-2 和Bax 表达情况HPH 组缺氧3 d Bcl-2 表达低于对照组(P<0.05),缺氧7、10、14 d 差异无统计学意义(P>0.05)。HSP70+HPH 组在各观察时间点Bcl-2 表达均高于HPH 组(P<0.05);KNK437+HSP70+HPH 组Bcl-2 表达缺氧3、7、10 d 低于HSP70+HPH 组(P<0.05),高于HPH 组但差异无统计学意义(P>0.05)。见图4、表3。

表3 各组新生大鼠肺组织Bcl-2-IOD/Area及单因素方差分析结果
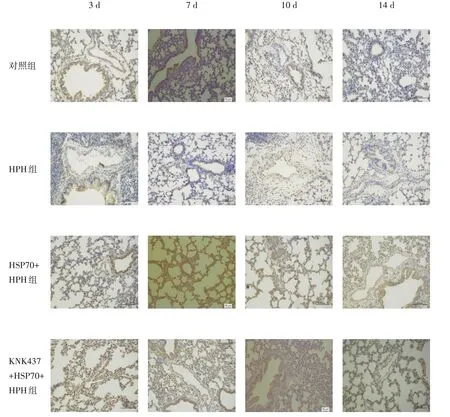
图4 各组新生大鼠肺组织中Bcl-2免疫组化染色(×200)
HPH 组缺氧3、7 d Bax 表达高于对照组(P<0.05),缺氧10、14 d 差异无统计学意义(P>0.05)。 HSP70+HPH 组缺氧3 d Bax 表达低于HPH 组(P<0.05),缺氧7 d 高于HPH 组(P<0.05),缺氧10、14 d 差异无统计学意义(P>0.05);KNK437+HSP70+HPH 组缺氧3 d Bax 表达高于HSP70+HPH 组(P<0.05),缺氧7 d 低于HSP70+HPH 组(P<0.05),缺氧10、14 d差异无统计学意义(P>0.05),各观察时间点与HPH组差异均无统计学意义(P>0.05)。见图5、表4。

表4 各组新生大鼠肺组织Bax-IOD/Area及单因素方差分析结果

图5 各组新生大鼠肺组织中Bax免疫组化染色(×200)
3 讨论
新生儿HPH 发病率逐年增多,病死率高[7],是威胁新生儿健康的重要疾病之一,目前尚缺乏切实有效阻断疾病进程的治疗手段[8-9]。已有研究证实,肺血管内皮细胞凋亡在成人HPH 的早期发生和后期进展中起着关键的作用[10]。 有研究发现肿瘤细胞HSP70 调控Bcl-2 和Bax 并参与了肿瘤细胞凋亡过程[15]。本研究发现经气管途径注入Ad-HSP70-EGFP转染液,提高了新生大鼠肺组织中HSP70 的表达,在缺氧早期促进了Bcl-2表达、抑制了Bax表达,抑制肺血管内皮细胞凋亡,降低了肺动脉压力。
经典转染腺病毒的途径是经尾静脉注射,但本实验中新生大鼠日龄和体重小,故经尾静脉穿刺难度大,成功率低,影响实验的顺利进行;结合气管插管在新生儿窒息复苏中迅速有效的应用,本研究采用经口气管插管途径将Ad-HSP70-EGFP 和Ad-CON-EGFP 经口气管内注入新生大鼠肺组织,荧光显微镜下观察EGFP 绿色荧光定位于细支气管周围处,继续观察发现随时间延长腺病毒绿色荧光逐渐减弱,于缺氧14 d 几乎观察不到绿色荧光;同时免疫组织化学结果显示,缺氧3、7 d,HSP70+HPH 组新生大鼠肺组织HSP70表达水平高于HPH组,但缺氧10、14 d两组间HSP70的表达水平差异不显著,说明经口气管插管能够将携带HSP70 的腺病毒成功转染至新大鼠肺组织,使HSP70 在新生大鼠肺组织中高表达,但随着时间延长腺病毒有衰减趋势,肺组织内HSP70水平也降低,考虑其原因可能是:腺病毒载体转染细胞是瞬时的,外源插入的基因片段只能转录却无法复制,不会由于细胞增殖而扩增,且HSP70mRNA 在细胞中半衰期短,易降解,从而导致外源引入的HSP70 绝对高表达无法实现。此外,HPH 组HSP70表达高于对照组,低于HSP70+HPH 组,说明缺氧激活少量内源性HSP70 表达,是机体抵抗缺氧应激刺激的保护机制之一。
本研究发现各观察时间点HPH 组平均RVSP 均高于对照组,表明新生大鼠HPH 模型构建成功。通过电镜观察肺血管内皮细胞发现,HPH 组缺氧3 d 即可观察到细胞凋亡变化,缺氧7d、10d 仍存在凋亡改变但程度下降,至缺氧14 d 即可观察到明显胶原纤维增生改变,说明在HPH 缺氧早期即发生肺血管内皮细胞凋亡,而随着缺氧时间延长,凋亡程度降低,而逐渐表现为增殖占据主导地位,此发现与成人HPH 中肺血管内皮细胞改变相似[10]。本研究发现随着缺氧时间延长HSP70 有衰减趋势,肺血管内皮细胞逐渐出现增殖变化,其中缺氧14 d HSP70+HPH 组增殖改变不如HPH 组明显,考虑腺病毒介导的HSP70 可在缺氧早期通过减少肺血管内皮细胞的凋亡发挥保护作用,从而保护后期肺血管内皮细胞表型损伤。同时研究发现HSP70 能够降低肺动脉压力,这与前期课题组发现一致[11],但HSP70 随着腺病毒衰减表达降低,其平均RVSP 仍低于同日龄HPH组,提示HSP70 通过早期抑制肺血管内皮细胞的凋亡发挥降低肺动脉压力的保护作用。
Bcl-2家族中的Bcl-2、Bax 基因是目前已知的细胞凋亡中最重要的调控基因,Bax 促进细胞凋亡,Bcl-2 抑制细胞凋亡,两者的失衡诱导细胞发生凋亡[12]。本研究发现HPH 组缺氧3 d Bcl-2 表达下降,缺氧3、7 d Bax 表达升高,结合电镜下缺氧3 d观察到凋亡改变,说明在缺氧早期抑凋亡蛋白Bcl-2 下降和凋亡蛋白Bax 升高促进了新生大鼠HPH 肺血管内皮细胞的凋亡,肺血管内皮细胞受到缺氧刺激,HSP70表达迅速升高,细胞受损严重超过HSP70 保护能力,细胞凋亡增加。而HSP70+HPH 组观察到Bcl-2 表达升高,缺氧3 d Bax 表达下降,HSP70 抑制剂组与HSP70组表达相反,证实腺病毒介导的HSP70可促进Bcl-2 表达上升、抑制Bax 表达下降,同时HSP70+HPH 组可降低肺动脉压力和延迟凋亡的发生,提示新生大鼠HPH 缺氧早期腺病毒介导HSP70可通过上调Bcl-2、下调Bax 表达,使内皮细胞凋亡改善,从而降低肺动脉压力。本研究显示缺氧后期Bcl-2 和Bax表达均下降,与文献[13]结果不一致,考虑Bcl-2 可能也参与晚期增殖的发生,但在新生大鼠HPH 缺氧后期可能不是影响肺血管内皮细胞增殖的主要因子,同时随着肺血管内皮细胞增殖的发生是否有部分Bcl-2 被消耗,本实验未阐述清楚,目前课题组正在进行该疾病增殖相关研究。
综上,本研究表明,新生大鼠HPH 经口气管插管腺病毒介导HSP70通过上调Bcl-2和下调Bax在新生大鼠HPH 缺氧早期减少肺血管内皮细胞凋亡发挥保护作用,从而降低肺动脉压力,有望成为治疗HPH 潜在靶点的作用。
猜你喜欢
承德医学院学报(2022年1期)2022-11-23
体育科技文献通报(2022年4期)2022-10-21
医学理论与实践(2022年14期)2022-08-01
传染病信息(2022年3期)2022-07-15
云南医药(2022年3期)2022-06-17
中国眼耳鼻喉科杂志(2022年3期)2022-05-29
中国动物保健(2022年2期)2022-05-05
中国循证心血管医学杂志(2022年1期)2022-03-15
安徽医学(2021年10期)2021-11-22
科学大观园(2021年1期)2021-01-11
