
基于微滴式数字PCR技术对猪内源逆转录病毒拷贝数的检测方法的建立及应用
2021-10-20 02:48:22卢天宇高虹杨博超于佳楠徐亚新王若琳秦
中国比较医学杂志 2021年9期
卢天宇高 虹杨博超于佳楠徐亚新王若琳秦 川
(中国医学科学院医学实验动物研究所,北京协和医学院比较医学中心,国家卫生健康委员会人类疾病比较医学重点实验室,国家中医药管理局人类疾病动物模型三级实验室,北京 100021)
在临床实践中,可供移植用的人类组织器官目前仍面临严重的短缺问题,使用源自猪的细胞、组织或器官进行异种移植,是解决该问题的潜在途径之一[1]。猪内源逆转录病毒(porcine endogenous retrovirus,PERV)是一种C型逆转录病毒,在进化过程中以前病毒DNA的形式被整合在猪基因组中[2]。目前PERV对于体内人类细胞的感染风险尚不确定,而供体猪不能通过无特定病原体培育、早期断奶或胚胎移植等方式消除PERV,从而造成了异种移植的潜在生物安全隐患[3]。因此,精确的测定PERV在供体动物中的拷贝数,以预测PERV的感染风险,对于异种移植的临床研究及应用具有重要的意义。
传统检测PERV拷贝数通常使用Southern blot[4]、荧光原位杂交[5]以及定量PCR(quantitative PCR,qPCR)[6-7]等方法,不同的方法对PERV的拷贝数测定结果有所差异[8]。其中,最常使用的qPCR方法在每次检测时都必须进行标准曲线的制备,而单次所能够检测样品数较少,加之PERV在不同来源的猪基因组中的拷贝数变化较大[9],扩增效率很难保证一致,易造成检测结果的准确性低和重复性差。
数字PCR作为新兴的技术,越来越广泛的用于核酸样品的分析,例如基因表达的检测[10]、病毒拷贝数的分析和绝对定量分析[11-13]以及拷贝数变异分析[14]等。相比于qPCR技术,使用数字PCR技术进行绝对定量分析不依赖于引物扩增效率,不需要使用标准品,可以直接测定出样品中目标基因的拷贝数,极大地减少了技术干扰,是真正意义上的绝对定量[15]。而微滴式数字PCR(droplet digital PCR,ddPCR)则是通过微滴发生油将整个反应体系分割成多个反应微滴,再在单个微滴内进行独立的PCR扩增,通过读取荧光信号确定阳性微滴和阴性微滴,根据泊松分布的原理推算目标基因的拷贝数[16-17]。
本研究采用双重荧光TaqMan探针,分别以猪GAPDH和TFRC作为内参基因,通过对于退火温度、反应循环数以及检测样本量等参数进行了系统的优化,建立了基于ddPCR技术的PERV拷贝数检测方法。通过与基于qPCR检测方法的比较,我们展示了所建立的PERV拷贝数检测新方法具有更高的灵敏度和更好重复性,为异种移植研究中PERV拷贝数的检测提供更好的技术和参考。
1 材料和方法
1.1 实验材料
1.1.1 质粒标准品
将猪GAPDH基因(3430~4294 bp)、TFRC基因(1200~1947 bp)和PERV的pol基因(3344~4115 bp),长度分别为730、665和748 bp的DNA片段,克隆到pEASY(3929 bp)载体上,获得pEASYGAPDH、pEASY-TFRC和pEASY-pol质粒。紫外分光光度计测定标准品质粒的浓度而计算拷贝数,通过10倍梯度稀释,获得107、106、105、104、103、102、10和1 copies/μL的浓度梯度稀释的质粒标准品。
1.1.2 猪基因组DNA
本研究中采用的猪基因组DNA样品,分别是来自2个猪胚胎成纤维细胞(porcine embryonic fibroblasts,PEF)、猪的肾表皮细胞系PK15以及来自3个不同个体的巴马小型猪的肺部组织,按照基因组提取试剂盒说明书获得细胞和组织基因组。
1.2 主要试剂与仪器
基因组提取试剂盒(全式金,中国);用于qPCR的TB Green Mix(TaKaRa公司,中国);微滴生成油、微滴读取油、ddPCR的反应液、微滴生成卡(Bio-Rad,美国);MseI内切酶(NEB,中国);96孔板(ABI,美国);Nano Drop紫外分光光度(赛默飞,美国);QX20TM Droplet Digital PCR系统(Bio-Rad,美国,包括微滴发生仪、微滴分析仪和热膜封口仪);CFX定量PCR仪(Bio-Rad,美国);T100梯度PCR仪(Bio-Rad,美国);引物和探针均由北京六合华大合成,具体信息见表1。

表1 检测PERV拷贝数所用的引物和探针序列Table 1 Sequences of primers and probes
1.3 实验方法
1.3.1 微滴数字PCR
(1)基因组酶切:500 ng基因组采用5 U MseI限制性内切酶,37℃水浴2 h。
(2)微滴生成:配制20μL反应体系(其中包含反应液mix 10μL,引物各900 nmol/L,探针各250 nmol/L,标准质粒或MseI酶切后的基因组1μL,补充水至20μL)加入微滴反应卡中。在微滴反应卡加入70μL微滴生成油,发生卡胶垫盖好后,将微滴发生卡放在微滴生成仪中生成微滴。
(3)PCR扩增:将生成的微滴转移到96孔板中,用PX1进行膜热封后,96孔板放在PCR仪上进行扩增。反应参为95℃10 min;94℃30 s,退火温度1 min,40~50个循环;98℃10 min,所有步骤升温速度均为2℃/s。
(4)微滴读取:扩增反应结束后,将96孔板放入QX200微滴分析仪中,采用Quantsoft软件进行微滴读取。
1.3.2 qPCR反应体系和条件
本实验中qPCR的反应体系为25μL(TB Green mix 12.5μL、10μmol/L PCR引物各1μL、模板DNA约100 ng、补充水至25μL)。PCR反应条件为95℃30 s;95℃5 s,60℃30 s,40个循环。
1.3.3 ddPCR退火温度的优化
选择63.0℃、61.9℃、60.9℃、59.9℃、58.6℃、57.5℃6个温度,进行退火和延伸温度的优化。以104copies/μL的3个质粒标准品以及1.25 ng的PEF 10#基因组为模板,确定最佳退火温度。
1.4 统计学方法
使用Quantasoft软件对ddPCR数据进行图像处理和结果分析,本研究中所有涉及的ddPCR反应微滴生成数均在10000~15000,符合ddPCR泊松分布的统计学原理,保证了后续分析结果的准确性。采用GraphPad进行数据统计和差异分析,结果用平均数±标准差(±s)表示,三组间比较采用单因素方差分析,以P<0.05为差异有显著性。
2 结果
2.1 微滴数字PCR退火温度的优化
以104copies/μL的标准品为模板,分别在63.0℃、61.9℃、60.9℃、59.9℃、58.6℃、57.5℃6个退火温度下进行PCR反应,结果显示pEASYGAPDH、pEASY-TFRC和pEASY-pol标准品在这6个温度条件下均能检测到荧光信号,在不同退火温度下没有显著差别(图1A和表2)。
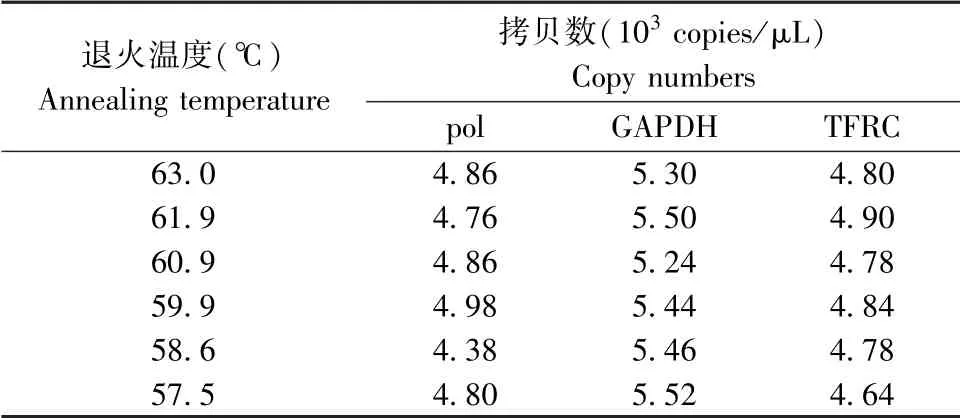
表2 标准品质粒在不同退火温度下ddPCR检测的拷贝数Table 2 Copy numbers of standard plasmids detected by ddPCR at different annealing temperatures
以63.0℃、61.9℃、60.9℃、59.9℃、58.6℃、57.5℃为退火和延伸温度,PEF基因组作为模板,分别以GAPDH和TFRC为内参基因进行ddPCR检测。如一维散点图所示,pol基因、GAPDH基因和TFRC基 因 分 别 在59.9℃~61.9℃、57.5℃~63.0℃、57.5℃~60.9℃进行退火和延伸温度适合(图1B和1C)。综上,本研究选用59.9℃作为后续ddPCR反应的最佳退火和延伸温度。
2.2 微滴数字PCR循环数的优化
根据Bio-Rad官方建议,ddPCR反应扩增循环数应为40个,但在以基因组为模板对退火温度优化时,PERVpol基因扩增后的阴、阳微滴较难分开(图1B和1C),易造成结果分析的偏差。因此,本研究通过提高PCR扩增循环数来优化这一反应条件。实验分别采用40、45和50个扩增循环对PEF的基因组模板进行PCR扩增。结果显示,扩增循环数的提高对于微滴生成并没有显著影响(图2A)。但是,随着扩增循环数的增加,pol基因扩增微滴的阴、阳性信号逐渐分开(图2B)。因此,本研究确定50个扩增循环作为后续ddPCR检测的反应条件。
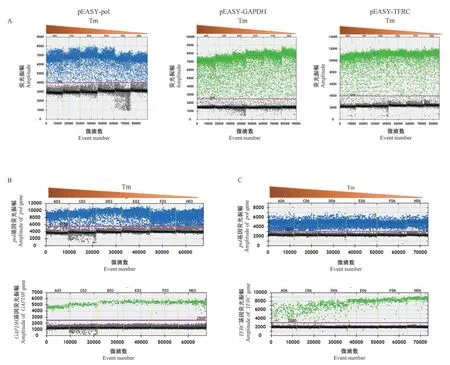
图1 不同退火温度下ddPCR扩增一维散点图Figure 1 One-dimensional scatter plot of ddPCR at different annealing temperatures
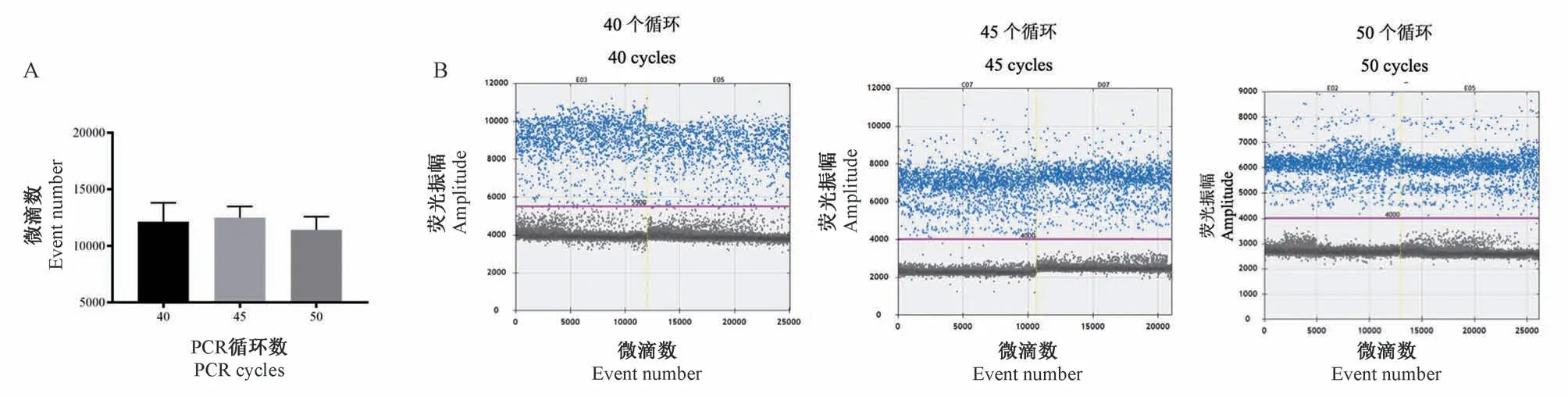
图2 ddPCR反应循环数优化Figure 2 Optimization of ddPCR cycle number
2.3 dd PCR检测范围的确定
本研究将3个基因的标准品质粒进行10倍浓度梯度稀释,对不同浓度的标准品分别进行ddPCR检测。结果显示,随着标准品浓度降低,阳性微滴数减少,3个基因的标准品以106copies/μL为模板进行PCR扩增产生的微滴全部为阳性微滴,超出了检测上限;以1 copy/μL的标准品为模板进行PCR扩增后,所产生的微滴中并未检测到阳性微滴(图3A)。采用ddPCR检测标准品的拷贝数与紫外分光光度计测定的DNA拷贝数,在检测范围内具有良好的相关性,pol基因r2=0.992,P<0.001、GAPDH基因r2=0.9976,P<0.0001、TFRC基因r2=0.996,P<0.001(图3B)。
本研究将PEF基因组按照107.5、5、2.5、1.25和0.625 ng为模板进行ddPCR。样本以10 ng基因组为模板,阳性微滴数超过了总微滴数的90%(图3C)。随着基因组模板浓度的降低,检测结果的变异系数提高(图3 D)。因此在满足ddPCR反应不超过检测上限的基础上,提高模板量能够提高结果的准确性。检测PERV拷贝数时,采用基因组模板在1.25~7.5 ng进行PERV拷贝数的检测为宜。
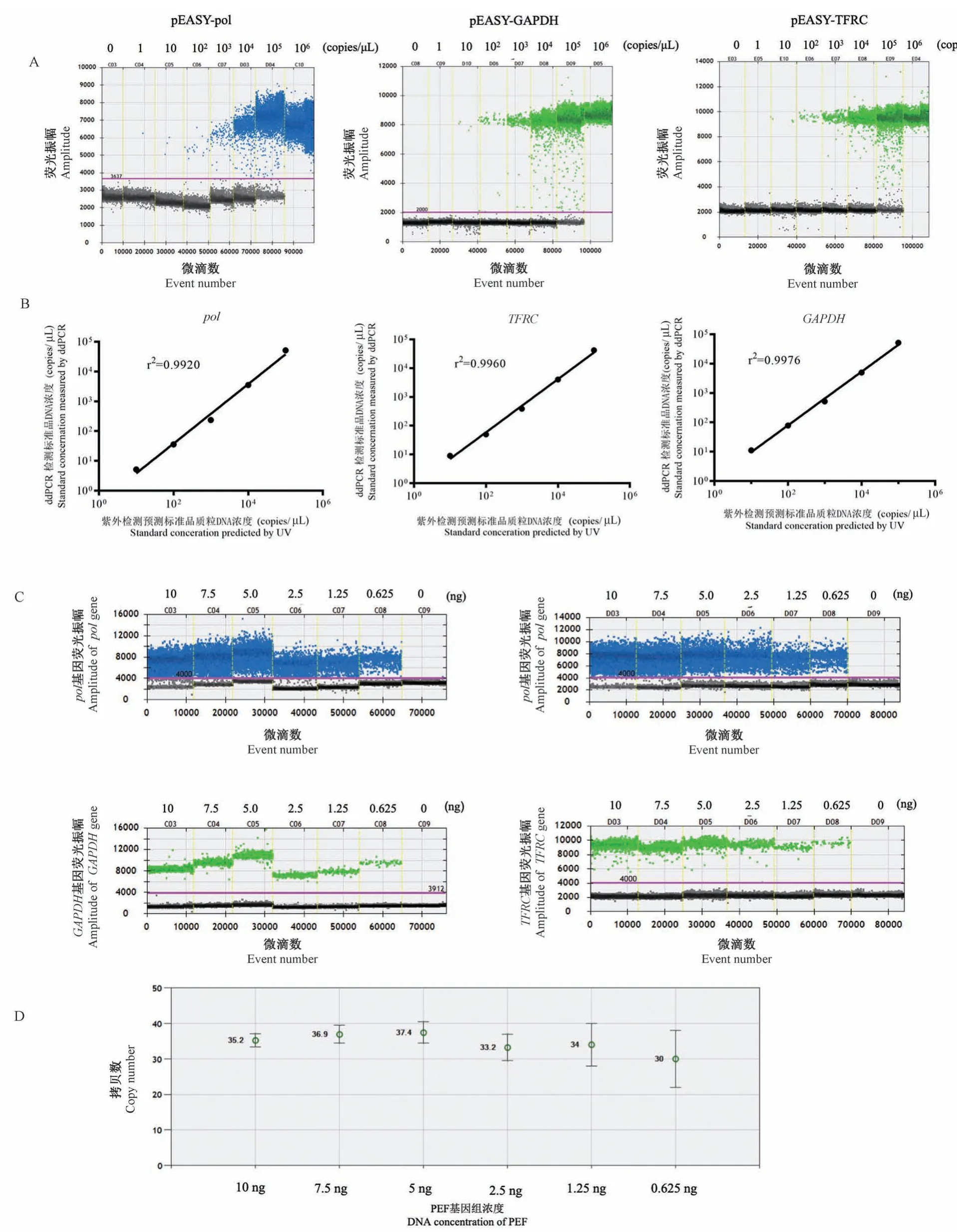
图3 ddPCR对于PERV拷贝数的检测范围Figure 3 Range of template for PERV copy number detected by ddPCR
2.4 样本检测
对来自3个细胞和3个肺组织的基因组样本,采用ddPCR和qPCR两种方法进行PERV拷贝数的检测。结果显示,ddPCR检测PERV拷贝数结果的变异系数在0.44%~8.29%之间,显著低于同一样本qPCR检测结果的变异系数,显示了ddPCR检测结果具有良好的稳定性(表3)。
表3 ddPCR和qPCR测定不同来源猪基因组中PERV的拷贝数(±s)Table 3 PERV copy numbers in pig genome detected by ddPCR and qPCR
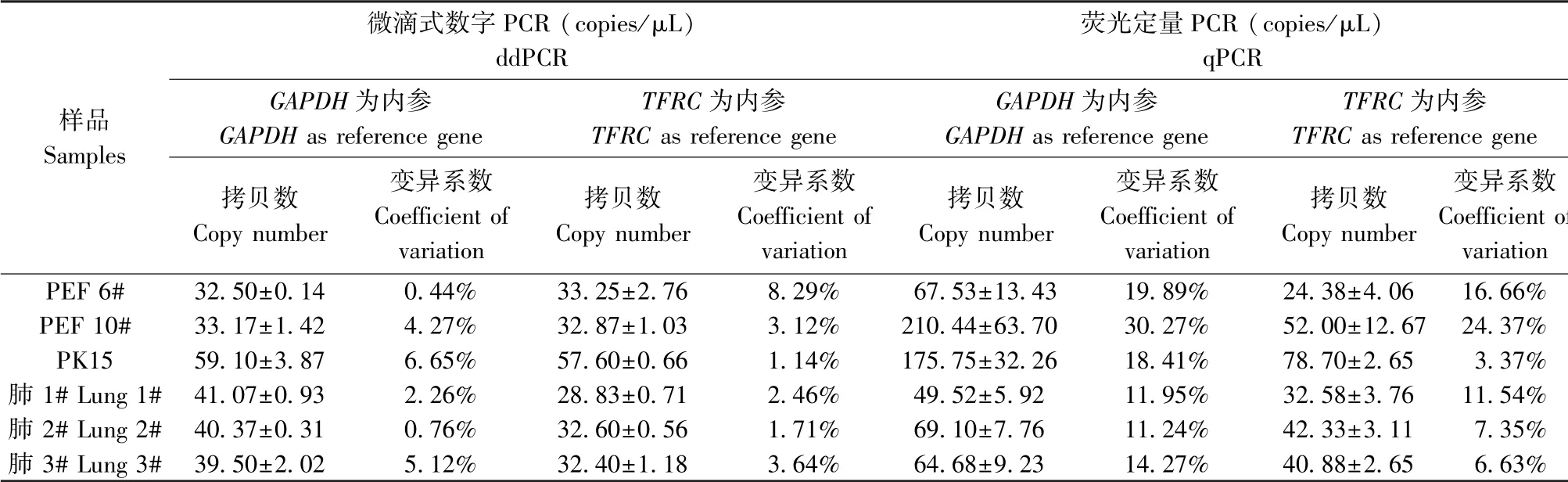
表3 ddPCR和qPCR测定不同来源猪基因组中PERV的拷贝数(±s)Table 3 PERV copy numbers in pig genome detected by ddPCR and qPCR
注:数据来自3次独立重复试验。Note.Data came from three independent replicates.
微滴式数字PCR(copies/μL)ddPCR样品Samples荧光定量PCR(copies/μL)qPCR GAPDH为内参GAPDH as reference gene TFRC为内参TFRC as reference gene GAPDH为内参GAPDH as reference gene TFRC为内参TFRC as reference gene拷贝数Copy number变异系数Coefficient of variation拷贝数Copy number变异系数Coefficient of variation拷贝数Copy number变异系数Coefficient of variation拷贝数Copy number变异系数Coefficient o variation PEF 6# 32.50±0.14 0.44% 33.25±2.76 8.29% 67.53±13.43 19.89% 24.38±4.06 16.66%PEF 10# 33.17±1.42 4.27% 32.87±1.03 3.12% 210.44±63.70 30.27% 52.00±12.67 24.37%PK15 59.10±3.87 6.65% 57.60±0.66 1.14% 175.75±32.26 18.41% 78.70±2.65 3.37%肺1#Lung 1# 41.07±0.93 2.26% 28.83±0.71 2.46% 49.52±5.92 11.95% 32.58±3.76 11.54%肺2#Lung 2# 40.37±0.31 0.76% 32.60±0.56 1.71% 69.10±7.76 11.24% 42.33±3.11 7.35%肺3#Lung 3# 39.50±2.02 5.12% 32.40±1.18 3.64% 64.68±9.23 14.27% 40.88±2.65 6.63%
3 讨论
随着免疫抑制药物研究和基因修饰供体动物的快速发展,将猪源器官异种移植到非人灵长类动物模型后,移植物可以存活数月甚至数年[19-21],异种移植是否可应用于临床也被不断的讨论。但是,体外研究发现PERV可以感染人源细胞[22-23],因此猪源组织器官异种移植到人体存在生物安全风险。从当前猪到非人灵长类模型的的研究结果,对供体猪体内PERV活性进行实时监测可以提前预测并防范该风险[24]。因此,对PERV拷贝的精确检测对于异种移植的临床应用具有重要的意义。
本研究采用ddPCR技术对猪基因组中PERV拷贝数的进行检测。与传统的qPCR方法相比,检测结果具有更好的稳定性和重复性,所需的被检测样本量也仅是qPCR方法所需的1/10~1/20,更适操作中,微滴生成后,需将微滴转移到其他模块中进行扩增和检测。在转移过程中可能造成液体的损失,从而导致目标分子定量的低估[25]。因此,本研究中我们采用TaqMan荧光探针双重微滴的方法,在同一微滴内对内参基因和目标基因进行绝对定量,从而规避了ddPCR的这一缺点。其次,ddPCR结果分析时确保阴性和阳性微滴的划分对于结果的准确性也非常重要,保证引物及探针的序列特异性和控制模板检测范围是控制这一因素的重要条件[26]。但是,PERV是作为前病毒被插入在猪基因组中,即使是高度保守的pol基因也含有点突变,且基因组中PERV的拷贝数较高,因此在ddPCR结果分析过程中微滴出现了“下雨”现象。为了解决这一问题,本研究中在选择特异性较好的引物和探针的条件下,还提高了PCR的循环数,从而提高了阳性微滴荧光的振幅,解决了阳性微滴的识别,提高了结果的准确性。这一结果也为采用ddPCR技术进行拷贝数变异分析时,优化检测条件提供了可行的方向。
在ddPCR中选择适合的内参基因,对于检测结果的准确性和稳定性具有重要的影响。GAPDH和TFRC基因是猪基因组中的单拷贝基因[27],在本研究中被选做内参基因。结果显示,在3个细胞来源的样品中,分别采用两个不同的内参基因,PERV拷贝数检测显示了良好的一致性,在后续的研究中可以作为PERV拷贝数检测的候选内参基因。但是在组织样品检测中,GAPDH和TFRC分别作为内参基因后所得到的结果略有差异。我们推测可能是由于组织样本中含有多种类型的细胞,尤其是存在大量血细胞,而造成了这一差异[8]。因此,在后续研究中,从组织样本分离单一类型的细胞进行PERV拷贝数的检测有可能提高检测结果的准确性。
综上所述,本研究建立了基于ddPCR技术对PERV拷贝数的检测方法,为异种移植供体动物中PERV的监测提供了可靠的方法,对于异种移植的临床研究具有重要的意义。
猜你喜欢
电子元件与材料(2022年1期)2022-02-14 02:55:34
郑州大学学报(工学版)(2022年1期)2022-01-17 01:58:16
纺织学报(2021年7期)2021-07-26 10:04:56
中国光学(2019年4期)2019-09-02 07:46:46
传媒评论(2019年12期)2019-08-24 07:55:10
传媒评论(2017年3期)2017-06-13 09:18:10
动画大王(漫画行)(2016年7期)2016-07-30 01:27:07
动画大王(漫画行)(2016年5期)2016-07-29 11:51:01
动画大王(漫画行)(2016年4期)2016-07-29 11:16:12
动画大王(漫画行)(2016年1期)2016-07-29 04:30:45
