
伴有先天性膈疝的Rubinstein⁃Taybi综合征1例报告
2021-10-19 07:34:02孔桂萍刘志峰郑必霞闫坤龙
南京医科大学学报(自然科学版) 2021年8期
孔桂萍,刘志峰,金 玉,郑必霞,闫坤龙*
1南京医科大学附属儿童医院消化科,2儿科重点实验室,江苏 南京 210008
Rubinstein⁃Taybi 综合征(Rubinstein⁃Taybi syn⁃drome,RSTS)是一种非常罕见的常染色体显性遗传病,1963 年由Jack Rubinstein 和Hooting Taybi 首次报道[1],发病率约为1∶125 000[2]。该病主要表现为智力低下、生长发育落后,宽而扁的拇指及第一脚趾以及特殊面容,包括高眉弓、长睫毛、眼裂下斜、钩状鼻、小下颚等[3]。本文对临床上高度怀疑该综合征的1例患儿采用二代测序技术进行相关基因检测,发现CREBBP基因突变后,用Sanger测序法验证患儿父母是否有该类型突变,并对临床表现和基因型进行分析,现报告如下。
1 病例资料
患儿,男,4月龄,2014年11月因“发热4 d,咳嗽3 d”于我院就诊。患儿为G1P1,41周顺产娩出,生时羊水清亮,新生儿Apgar评分9分,出生体重3.3 kg,身长47 cm。出生即发现头发、眉毛、睫毛黑而浓密,宽而扁的拇指。生后体重增长慢,目前不能注视追人和与人眼神交流,不能抬头,双手不会主动拿物。父母体健,非近亲婚配。否认家族中有类似疾病患者。既往因先天性隔疝、反复肺部感染多次住院治疗,每次感染后血象均增高明显[白细胞(19~35)×10-9/L]。
入院查体:体温37.2 ℃,脉搏132 次/min,呼吸42次/min,体重7 kg,身高58 cm(低于正常同龄男童参考值第3百分位点),上部量36 cm,下部量22 cm,指间距49 cm,头围36.5 cm(低于正常同龄男童参考值第3百分位点),前囟平软1.5 cm×1.5 cm。头发浓密,发丝粗黑,质硬,前后发际低,眉毛粗长,色黑,高拱形眉弓;双内眦赘皮,双眼裂下斜,鼻外形正常;口唇红润,腭弓高,牙齿未萌出。耳位低。心肺检查未见异常,腹部稍膨隆,肝脏肋下2 cm,质软,脾脏肋下未触及。双手无通贯掌,双拇指及拇趾宽;阴茎发育正常,双侧睾丸触及。肌力及肌张力偏低,胸骨、肋骨和脊柱正常,病理征未引出(图1)。入院诊断为:支气管肺炎、先天性隔疝、精神运动发育迟滞。头颅CT、MRI、体液免疫、细胞免疫、血氨基酸及尿有机酸代谢筛查均无明显异常。
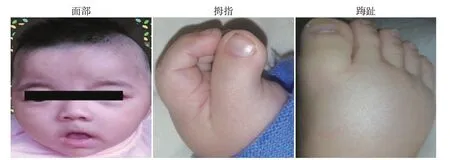
图1 患儿临床表现
5 月龄时(2014 年12 月)因先天性隔疝于我院胸外科行隔疝修补术。7月龄时行智能发育量表评分,智力发育指数(MDI)、运动发育指数(PDI)均<50,评价相当于4.4月龄。
征得父母知情同意后,取患儿及其父母外周静脉抗凝血2 mL,基因全外显子测序由北京智因东方转化医学研究中心协助完成。使用BloodGen Midi Kit(北京康为世纪生物科技有限公司)提取患者全基因组DNA,构建文库,应用Illumina Hiseq XTen 平台对全基因组进行高通量测序,利用Illumina 官方basecall 分析软件BclToFastq 得到原始数据(raw data)。测序数据运用BWA 软件与UCSC 数据库提供的人类基因组hg19 参考序列进行比对。根据测序深度,突变质量,对检测得到的单核苷酸多态性(SNP),插入或缺失(InDel)进行过滤筛选,得到高质量可靠的突变。使用SIFT利用基于同源比对,蛋白结构的保守性等的算法,预测筛选出的变异对蛋白质的影响。对明确或可能与受检者临床表型相关的基因突变,采用一代测序(Sanger法)验证。
经过测序,患儿存在CREBBP基因27号外显子上的复杂移码突变:c.4499至c.4493缺失TGCAGTC插入AAGCA(图2)。该突变在千人数据库、dbSNP数据库及hapmap 数据库中没有查到相关记载,缺失/插入突变的致病性明确,根据氨基酸的保守性、公共数据库资料和患儿典型临床表现,定义为致病突变,最终诊断为RSTS。其父母标本检测未发现有该基因位点的突变,患儿为de novo变异。
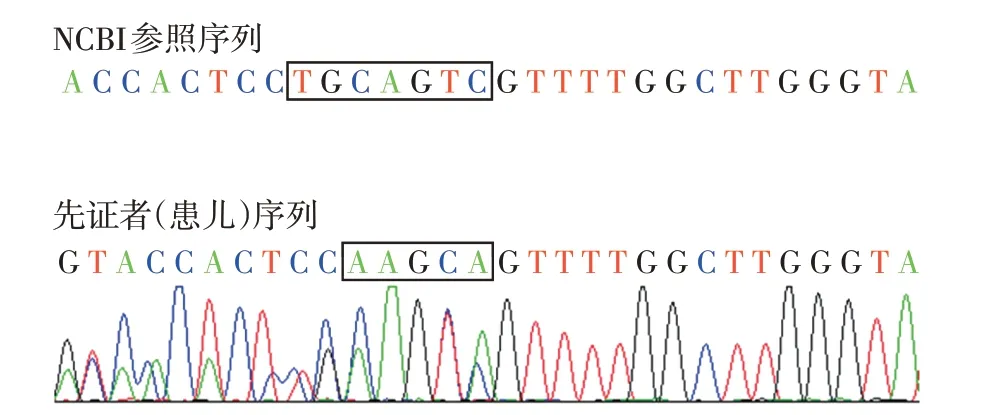
图2 基因测序图
2 讨论
作为多系统疾病,RSTS 的主要特征为3 类:颅面畸形、骨骼畸形以及生长和精神运动发育迟滞。此外,还可伴发其他表现,如眼睑下垂、白内障、青光眼,先天性心脏疾病,泌尿系统畸形、多毛、前额红斑、瘢痕皮肤、恶性肿瘤等[3-4]。婴儿期开始出现明显生长发育落后,通常低于同龄儿童参考值第15百分位水平。智商平均在35~50 分,但也有智商在正常低限的报道[5]。后期易出现注意力集中时间短、运动刻板印象、协调性差等行为异常[6],青春期会有焦虑,情绪不稳和攻击性行为等[3]。超过90%的病例可以存活到成年[7]。本文所述病例存在生长和精神运动发育落后、宽而扁的拇指和第一脚趾、特殊面容、多毛等表现,与RSTS 高度相符。该患儿同时还存在先天性隔疝,为该病的首次发现。
目前认为,55%~70%的RSTS 病例来源于CREBBP基因突变[8],5%~8%源于EP300 基因突变[5],其余病例原因尚未明确[9]。CREBBP 基因位于染色体16p13.3,其同源物EP300位于染色体22q13.2,两者均由相等数量的外显子(31)组成。RSTS 发病机制尚不明确,已知CREBBP广泛表达,具有固有的组蛋白乙酰转移酶(HAT)活性,可作为支架通过染色质重塑来稳定与转录复合物的其他蛋白质相互作用,从而调节许多影响细胞途径的基因表达[10]。CREBBP 变异可能导致CREB 结合蛋白被截短或被氨基酸取代,HAT 结构域中的致病变异会干扰组蛋白的乙酰化,这是转录激活的重要步骤。EP300 编码p300 转录激活蛋白,该蛋白与CREBBP 在氨基酸水平上具有63%的同源性,起HAT 的作用,通过染色质重塑调节转录,并在细胞增殖和分化中发挥重要作用。致病性变体导致P300 蛋白被截断或等位基因表达消失,这可能导致HAT 活性丧失[3-5]。本文所述病例发现CRBBP基因中一处新的移码突变,为首次报道,推测患儿存在先天性膈疝可能与该新变异位点有关,但两者关系仍有待进一步深入研究探讨。
CREBBP 的突变谱由点变异(30%~50%)和缺失(~10%)组成[9],EP300 突变患者表型被认为比CREBBP 突变的先证者要轻[5]。本例患儿为CREBBP基因复杂移码突变,临床表现典型,与文献报道一致。由于具有高度变异性,迄今为止,有关RSTS基因型与临床表现的关系尚无定论。
大多数RSTS病例报道为新发突变引起,仅有少数影响同胞的相关报道,垂直传输亦极为罕见[3]。如果先证者的父母受到影响和/或已知在先证者中鉴定出CREBBP 或EP300 致病变异,则同胞的风险为50%[7]。一旦确定了引起RSTS 的致病变体,就可以进行产前检测,虽然该病再发风险通常较低,但适当的遗传咨询对于产前诊断还是必要的。
本例基因变异位点在国内外属首次发现,临床表现伴有先天性隔疝亦属首次报道。RSTS 作为一种极为罕见、复杂的疾病,虽然已经明确了一些临床特征,但目前仍然存在许多未解决的问题,仍需要进行多中心研究,包括新生儿至成年阶段,以扩大对临床表型的认识,确定不同基因型与表型的相关性,从而有助于早期识别诊断,及时干预。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21 02:14:52
心肺血管病杂志(2020年5期)2021-01-14 00:43:30
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
科学生活(2019年7期)2020-01-01 08:28:02
养殖与饲料(2019年10期)2019-02-25 14:52:37
山东畜牧兽医(2018年3期)2018-04-26 09:10:34
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
百科知识(2015年18期)2015-09-10 07:22:44
中国当代医药(2015年30期)2015-03-01 02:08:19
