
固相萃取/超高效液相色谱-三重四极杆质谱法测定人参、人参叶与人参花中10种人参皂苷含量
2021-10-17 07:59:02陈树东胡文军孔祥词梁土金崔媛媛钟满霞
分析测试学报 2021年9期
陈树东,胡文军,孔祥词,冯 锐,梁土金,崔媛媛,钟满霞
(1.广东省中药研究所,广东 广州 510640;2.广东医科大学,广东 东莞 523808)
人参作为传统名贵中药材,具有大补元气、补脾益肺、生津、安神益智等功效[1]。人参皂苷作为人参的主要活性成分,在免疫调节、抗肿瘤、抗抑郁等神经系统调节方面具有一定的药理作用[2-3]。人参中分离鉴定了近百种人参皂苷类化合物,根据苷元结构的不同,可分为四环三萜类结构的达玛烷型的原人参二醇型皂苷(PPD)、原人参三醇型皂苷(PPT)和五环三萜结构的齐墩果烷型皂苷3种类型,其中,以原人参二醇型Rb1、Rb2、Rb3、Rc、Rd、Rg3和原人参三醇型Re、Rf、Rg1、Rg2等常见的人参皂苷为主要成分[4]。目前,人参皂苷的研究主要集中在人参根部,对于人参地上部分的报道较少,有研究表明人参叶[5-8]、人参花[9-11]均含有人参皂苷类成分,甚至作为人参种植副产物的人参叶、人参花中的功效成分和营养物质含量更高[12]。因此,有必要对人参不同部位的人参皂苷含量进行准确定量。
人参皂苷的测定方法主要包括高效液相色谱法(HPLC)[1,13-15]、液相色谱−质谱联用法[16-17]、胶束电动毛细管色谱法(MEKC)[18]等,其中,MEKC法由于仪器成本和实际应用的限制,较难普及。质谱法检测人参皂苷的多组分常采用四极杆和飞行时间质谱检测器(QTOF)[16-17],QTOF检测器具有高分辨率,适用于未知物质的定性鉴别,而三重四极杆质谱兼具灵敏度高、选择性强的优势,更适用于已知物质的定量分析;HPLC法是人参皂苷的主要检测方法,但由于人参皂苷在紫外区的吸收较弱,如果样品中的复杂成分未进行有效分离,在紫外吸收的临界波长检测人参皂苷时,基质中含有的大量杂峰会对检测产生较大干扰。本研究根据人参皂苷的化学性质,采用Alumina-N/XAD-2SPE Cartridge复合固相萃取柱针对性地分离和净化样品中的人参皂苷成分,结合超高效液相色谱−三重四极杆质谱建立了达玛烷型人参皂苷Rb1、Rb2、Rb3、Rc、Rd、Rg3、Re、Rf、Rg1和Rg2含量的检测方法,并对人参、人参花和人参叶中10种人参皂苷的含量和分布情况进行了初步研究,以期为进一步开发和利用人参资源,更好控制人参质量提供依据。
1 实验部分
1.1 试剂与仪器
甲醇、乙腈(色谱纯,德国Merck公司);乙醇(分析纯,广州化学试剂厂);甲酸、乙酸铵(LCMS级,上海安谱实验科技股份有限公司);正丁醇(分析纯,上海麦克林生化科技有限公司);固相萃取小柱(Alumina-N/XAD-2SPE Cartridge,1g/4g,10mL,上海安谱实验科技股份有限公司)。对照品:人参皂苷Re(批号110754-201827,下同)、Rg1(110703-201731)、Rb1(110704-201827)、Rg3(110804-201504)购于中国食品药品检定研究院,人参皂苷Rb2(Z99800205)、Rb3(Y8650010)、Rc(U9330025)、Rd(R7880050)、Rf(T7810020)、Rg2(47560010)购于上海安谱实验科技股份有限公司;人参、人参叶、人参花样品各3批,均为市售并经本实验室鉴别。
TSQ Quantum Access MAX三重四极杆质谱仪、UltiMate3000高效液相色谱仪(美国Thermo Scientific公司);Sartorius CPA225D电子天平、BSA224S-CW电子天平(德国Sartorius公司);5424R冷冻离心机(德国Eppendorf公司);MIX-1振荡涡旋混合器(上海托莫斯科学仪器有限公司);KQ-250E超声波水浴器(昆山市超声仪器有限公司);实验用水由Milli-Q Advantage A10超纯水系统(美国Millipore公司)制备。
1.2 实验条件
1.2.1 色谱条件 Hypersil Gold C18色谱柱(100mm×2.1 mm,1.9 µm);流动相:A为5mmol/L乙酸铵溶液(含0.1 %甲酸),B为乙腈。梯度洗脱条件:0~4.0 min,81%A;4.0 ~6.0 min,81%~79%A;6.0 ~8.0 min,79%~72% A;8.0 ~15.0 min,72%~69% A;15.0 ~20.0 min,69%~54% A;20.0 ~20.5 min,54%~10%A;20.5 ~22.0 min,10%A;22.0 ~22.5 min,10%~81%A;流速:0.4 mL/min;柱温:30℃;进样量:5µL。
1.2.2 质谱条件离子源:电喷雾离子源,负离子模式(ESI-);扫描方式:多反应监测(MRM);离子传输毛细管温度:350℃;电喷雾电压:3000V;蒸发温度:400℃;碰撞气体压力:0.2 Pa;辅助气:3.6 mL/min;鞘气:10.5 mL/min。10种分析物的监测离子对(Q1/Q3)、碰撞能量(CE)和管透镜补偿电压见表1。
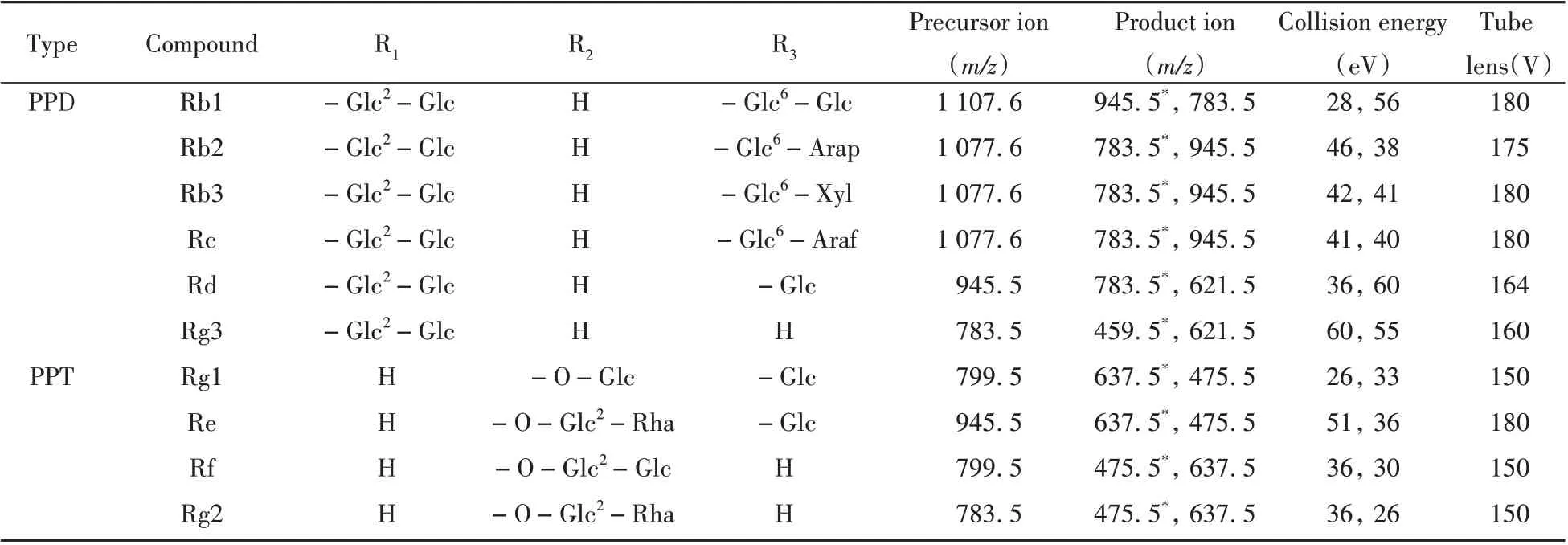
表1 10种目标化合物的化学结构和质谱参数Table1 Chemical structures and MS/MS parameters of ten analytes
1.3 实验方法
1.3.1 对照品溶液的制备 分别准确称取各目标物标准品0.01 g,用甲醇溶解并配制成1mg/mL的标准储备液,于-18℃保存。混合标准溶液:分别吸取一定量的标准储备液,用甲醇稀释,涡旋混匀,配制成质量浓度为10µg/mL的混合标准溶液,于-18℃保存。混合标准工作液:移取适量混合标准溶液,用甲醇-水(30∶70,体积比)配制成系列混合标准工作液,现用现配。
1.3.2 供试品溶液的制备提取:称取过四号筛的样品粉末0.2 g,精密加50mL水饱和的正丁醇,密塞,放置过夜,超声处理(功率250W,频率50kHz)30min,过滤,弃去初滤液,精密量取续滤液25mL,置蒸发皿中蒸干,残渣用10mL水溶解,待固相萃取净化。
净化:分别用20mL70%乙醇和20mL水对Alumina-N/XAD-2SPE Cartridge固相萃取柱进行活化,取5.0 mL样品提取溶液于已活化的SPE柱上样,分别用20mL水进行淋洗,用20mL70%乙醇进行洗脱,收集洗脱液;洗脱液用甲醇-水(30∶70)定容至25.0 mL,混匀后过0.22 µm滤膜,即得;当样品浓度超过标准曲线上限浓度时,采用倍量稀释将浓度控制在标准曲线范围内,然后测定。
2 结果与讨论
2.1 人参皂苷的质谱裂解过程及条件优化
人参皂苷的相关质谱研究表明,实验条件对电喷雾电离质谱的影响较大[19-20]。本文10种人参皂苷的分子结构均以相同的皂苷苷元和不同的糖基构成。通过对人参皂苷电离裂解产生的碎片进行分析发现,在ESI-模式下,原人参二醇型皂苷的准分子离子在碰撞裂解时,R3位置的糖基更易断裂,人参皂苷Rb1先后失去2个葡萄糖基(-Glc)形成m/z945.5 和783.5 两个特征离子峰(见表1);人参皂苷Rb2、Rb3、Rc首先断裂失去分子量相同的阿拉伯吡喃糖基(-Arap)、木糖基(-Xyl)和阿拉伯呋喃糖基(-Araf),形成m/z945.5 的特征离子峰,再进一步失去1个葡萄糖基,形成m/z783.5 的特征离子峰;人参皂苷Rd、Rg3在R3位置的糖基断裂后,通过调节碰撞能量使R1位置的葡萄糖基进一步断裂丢失,形成m/z621.5 和459.5 的特征离子峰。而原人参三醇型皂苷质谱的裂解过程也呈现相似的规律,通过分别在R3和R2位置断裂失去葡萄糖基和鼠李糖基(-Rha),先后形成m/z637.5 和475.5 的特征离子峰。可以看出,原人参二醇型皂苷和原人参三醇型皂苷的离子碎片呈现明显的规律性。
在确定一级质谱准分子离子峰和相应的碎片离子后,由于人参皂苷裂解所需的能量不同,实验通过对喷雾电压、碰撞能量和管透镜补偿电压等条件逐步进行优化,进一步提升目标物的灵敏度。后续研究中,在缺少对照品的情况下,根据人参皂苷在质谱中的裂解规律,可对其他人参皂苷的相对含量进行分析,也可对未知人参皂苷的结构进行推断和鉴定,进一步获取人参皂苷的苷元、糖基和相对分子量等信息。
2.2 色谱条件的优化
根据人参皂苷分子结构的碰撞断裂规律,进一步对色谱条件进行优化。首先通过对流动相的比较,发现以乙腈体系为流动相的洗脱能力优于甲醇体系,且系统平衡时间相对更快。在质谱条件优化过程中发现,乙腈-水流动相体系中加入甲酸可提高离子化效率,增强人参皂苷的响应信号和稳定性;另外,添加一定比例的乙酸铵可促进人参皂苷的离子化过程,稳定流动相的pH值,改善部分目标物质的峰形和提高洗脱能力;但噪音值同步增大,最终确定乙酸铵的最佳浓度为5mmol/L。最后,通过调节梯度洗脱程序使互为同分异构体的人参皂苷进一步分离,由目标物的结构可知,原人参二醇型皂苷Rc、Rb2和Rb3与原人参三醇型皂苷Rg1和Rf分别为两组同分异构体,虽然检测离子碎片相同,但可通过保留时间的不同得到分离(见图1);而另一组保留时间接近的人参皂苷Rg3和Rg2同分异构体,由于皂苷结构的不同,也能通过离子碎片的不同加以区分。在优化的色谱条件下,10种待测物质均得到有效分离,从而实现准确定量的目的。
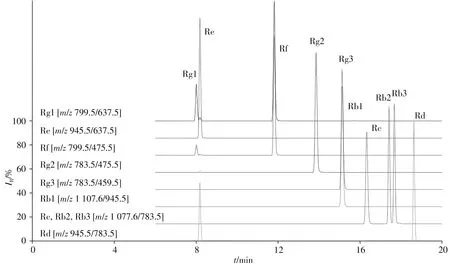
图1 10种人参皂苷的定量离子图谱Fig.1 Quantitative ion chromatograms of ten ginsenosides
2.3 固相萃取条件的优化
《中国药典》2020年版分别只收载了人参和人参叶中人参皂苷Re、Rg1、Rb1以及人参皂苷Rg1、Re的检测方法和质量指标[1],相关地方标准也未收载人参花的质量控制标准。在实际检测中,由于人参叶和人参花中含有多种皂苷和色素、氨基酸、糖类等成分,在常规液相色谱检测中无法准确进行定量。为进一步分离、净化目标物质,本研究选择Alumina-N/XAD-2SPE Cartridge复合固相萃取柱作为前处理的净化柱,柱填料分别为XAD-2大孔吸附树脂和中性氧化铝,通过XAD-2大孔树脂的分子排阻作用对样品中的糖类、蛋白质和有机酸等进行洗脱[21],通过中性氧化铝对样品中的色素、黄酮类物质进行吸附,从而使人参皂苷类物质得到进一步富集、净化。此外,还对提取溶剂、提取工艺和提取时间进行了优化:研究了不同比例的提取溶剂,发现以水饱和的正丁醇溶液为提取溶剂对人参皂苷的提取率最高;与回流法和索氏提取法相比,超声提取法简单、重复性好、效果好;粉碎后的样品分别用水饱和的正丁醇溶液提取10、20、30、40、50、60min,结果显示,30min后提取的人参皂苷含量基本不变,因此最佳超声提取时间为30min。优化的固相萃取条件如“1.3.2 ”所示。
2.4 基质效应的考察
选取人参、人参叶、人参花样品,分别比较固相萃取前、后3种基质对10种目标物质的影响,并以通过标准加入法计算供试品溶液与对照品溶液相应浓度点的峰面积比值表示基质效应因子(MF),MF值越接近1,表明基质效应越低。结果显示,10种目标化合物固相萃取净化前在人参、人参叶和人参花样品中的MF值分别为0.819 ~0.903 、0.757 ~0.896 、0.775 ~0.923 ;经固相萃取净化后的MF值分别为0.945 ~0.988 、0.925 ~0.971 、0.901 ~0.979 。说明样品经固相萃取的分离净化和稀释后,较好地消除了基质效应,保证了定性定量结果的准确、可靠。
2.5 方法学验证
2.5.1 线性关系、检出限及定量下限分别精密量取混合对照品溶液适量,用甲醇-水(30∶70)稀释,混匀制得系列混合对照品溶液。采用本方法进样测定,以系列混合对照品溶液的质量浓度为横坐标(X,ng/mL),质谱峰面积为纵坐标(Y),进行线性回归;并以线性方程中最低添加浓度的数据,分别根据3倍和10倍信噪比计算人参皂苷的检出限(LOD)与定量下限(LOQ)。结果表明,10种目标物质均在5~2500ng/mL范围内线性关系良好,相关系数(r)不小于0.9980 ;LOD和LOQ分别为0.25 ~2.5 mg/kg和0.75 ~7.5 mg/kg(见表2)。
2.5.2 方法回收率及相对标准偏差以人参叶为典型样品,分别取同一批次人参叶样品,添加适量的高、中、低浓度混合标准品溶液,每个水平做6个平行样,按上述供试品溶液制备方法和色谱质谱条件进行分析,计算各人参皂苷的回收率和相对标准偏差(RSD),结果见表2。结果表明,样品中10种人参皂苷的回收率为87.3 %~110%,RSD为1.4 %~9.3 %,说明该方法具有良好的准确度和精密度。
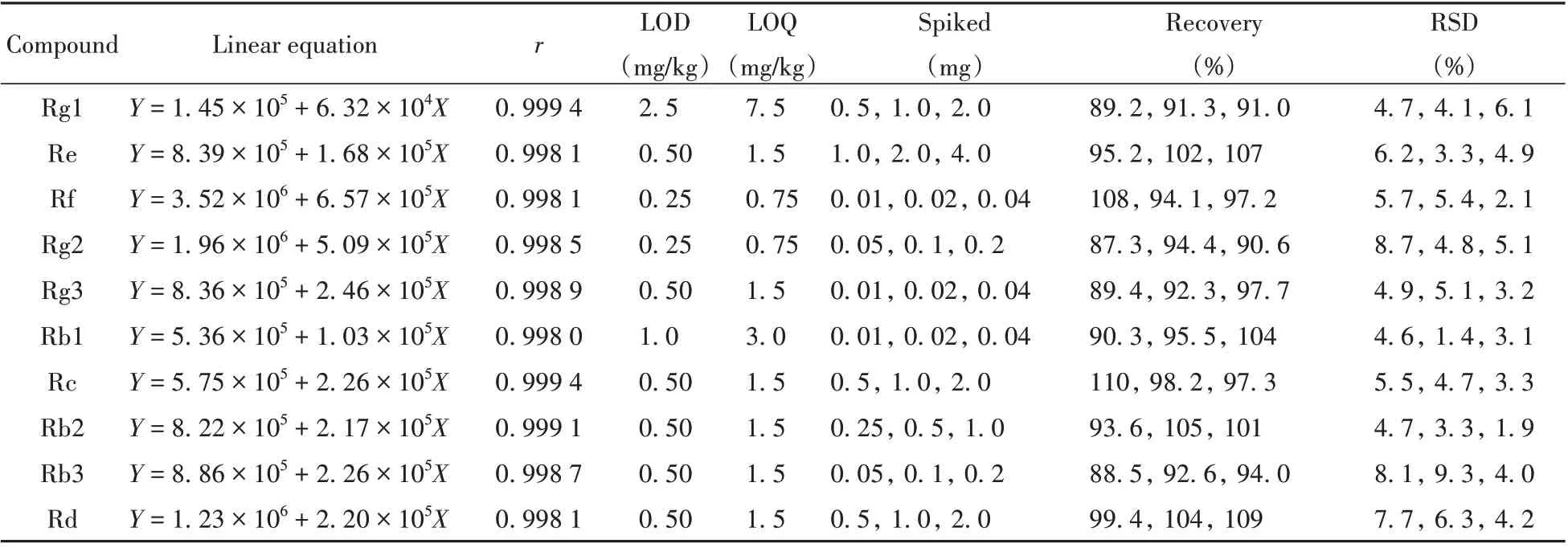
表2 10种人参皂苷的线性关系、检出限、定量下限、回收率及相对标准偏差Table2 Linear relations,LODs,LOQs,recoveries and RSDs of10analytes
2.5.3 稳定性取同一批次人参叶供试品溶液,分别于放置0、4、8、12、24h时进样测定,并计算相应的RSD。结果表明,在24h内10种人参皂苷的响应值RSD均小于5.5%,表明方法稳定性较好,满足日常检测需求。
2.6 实际样品的检测
分别取不同批次人参、人参叶和人参花样品各3批,按上述方法进行测定,结果见表3。结果显示,人参、人参叶和人参花样品均能检出10种人参皂苷,10种人参皂苷在人参根部的总量为13.44 ~20.11 mg/g,在人参叶中为56.83 ~59.81 mg/g,在人参花中为62.02 ~87.83 mg/g。除了人参皂苷Rf、Rb1外,另8种人参皂苷在人参叶和人参花中的含量更高,特别是人参皂苷Re、Rc和Rd,在人参叶和人参花中的含量显著高于人参根部。

表3 人参、人参叶和人参花样品中10种人参皂苷的含量(mg/g)Table3 Contents of10ginsenosides in roots,leaves and flowers of ginseng(mg/g)
3 结 论
人参的传统药用部位为根部,作为人参种植的副产物,人参叶和人参花大部分被遗弃,少部分以代用茶或化妆品原材料的形式被加以利用[22]。本文通过对人参与人参叶、花中的实际检测结果进行比较,发现人参叶、花所含成分与人参相似,且部分人参皂苷单体和10种主要人参皂苷总量均远大于人参根部。由于人参的根和根茎的生长周期较长,通过进一步科学、充分利用这些人参副产物,可以延长人参产业链,优化产业结构,同时扩大人参药材的使用范围,使药材资源利用最大化,并减少人参的需求量及森林植被和环境的破坏。
猜你喜欢
基层中医药(2021年7期)2021-11-02 07:20:06
食品安全导刊(2021年20期)2021-08-30 06:39:48
海峡姐妹(2019年8期)2019-09-03 01:01:04
中成药(2018年9期)2018-10-09 07:19:04
中成药(2017年9期)2017-12-19 13:34:40
中成药(2017年6期)2017-06-13 07:30:34
广东第二课堂·小学(2016年11期)2016-12-06 14:28:02
华人时刊(2016年13期)2016-04-05 05:50:15
当代化工研究(2016年5期)2016-03-20 16:21:35
中国药业(2014年16期)2014-05-14 06:46:20
