
黄芪中黄酮类化合物的超临界流体色谱分离方法研究
2021-10-17 07:58赵淑军董姣姣贾志鑫闫晓宁郁映婷陈奕君李月婷肖红斌
分析测试学报 2021年9期
赵淑军,董姣姣,刘 洁,贾志鑫,闫晓宁,郁映婷,陈奕君,李月婷,肖红斌*
(1.北京中医药大学 中药学院,北京 100029;2.北京中医药大学 中药分析与转化研究中心,北京 100029;3.北京中医药大学 北京中医药研究院,北京 100029)
黄芪为豆科植物蒙古黄芪Astragalus membranaceus(Fisch.)Bge.var.mongholicus(Bge.)Hsiao或膜荚黄芪Astragalus membranaceus(Fisch.)Bge.的干燥根,具有补气升阳、固表止汗、利水消肿、生津养血、行滞通痹和敛疮生肌等功效[1]。研究表明,黄芪主要含黄酮、皂苷及多糖三大类化合物,其中黄酮类化合物主要包括异黄酮类、异黄烷类、紫檀烷类等[2]。黄酮类化合物具有良好的药理活性,以及抗氧化、抗心肌缺血、抗炎、抗骨质疏松、促进细胞免疫等作用[3-4]。2010版《中国药典》已将黄芪中毛蕊异黄酮葡萄糖苷列为质量评价指标[5]。目前,分离测定黄芪中黄酮类成分的方法主要有薄层色谱法(TLC)[6]、高效液相色谱法(HPLC)[7-8]、高效液相色谱-高分辨飞行时间质谱法(HPLC-ESI-TOF MS)[9]等。TLC法具有快速简便的优势,但作为定量方法,具有损失样品和误差较大等缺点。张妍等[10]利用HPLC同时测定黄芪中10种黄酮类成分,用时54min,耗时较长;陈婷等[9]采用HPLC-ESITOF MS法测定黄芪中黄酮类成分,方法灵敏度高,能够同时定量分析24种黄酮类成分,时长为25min,但所需设备昂贵,不具有普遍性。
超临界流体色谱(SFC)被认为是一种能够与传统液相色谱法互为补充的快速分离技术,已收录在2015版《中国药典》新增通则中[11]。SFC的快速分析技术不仅适用于手性化合物的分析,在复杂成分中药中的应用也逐渐增加。Pfeifer等[12]采用SFC法在6min内成功分离8种香豆素类化合物,并测定了白芷中3种香豆素化合物的含量,而传统分析方法至少需要20min。Murauer等[13]采用SFC法在金鸡纳树中分析了7种结构极为相似的生物碱类化合物。Zhu等[14]建立的SFC法成功分离了20个螺甾皂苷,并总结了SFC分离甾体皂苷的色谱行为规律及特点,证明了SFC对区别皂苷中不同位置的取代基具有优势。Huang等[15]建立的SFC法在18min内实现了12种黄酮类化合物的基线分离,并在滁菊、贡菊、杭菊、亳菊中测定了5种黄酮类化合物的含量。
针对黄芪中黄酮类化学成分的快速分离与分析,本文以黄芪中9种黄酮类化合物(毛蕊异黄酮、芒柄花素、山奈酚、槲皮素、异鼠李素、毛蕊异黄酮葡萄糖苷、紫云英苷、异槲皮苷、芒柄花苷)为目标物,考察影响SFC法分离的各种因素,建立了快速分析方法,并与HPLC法进行比较。最终建立了测定黄芪饮片中5种主要黄酮类化合物含量的SFC方法,进而对黄芪饮片样品进行测定。
1 实验部分
1.1 仪器、试剂与试药
Agilent1260高效液相色谱仪、Agilent1260超临界流体色谱仪、Agilent ZORBAX RX-SIL色谱柱(4.6 mm×150mm,5µm)购于美国安捷伦公司;Waters ACQUITY UPC2TMBEH2-EP色谱柱(3mm×100mm,1.7 µm)购于沃特世科技(上海)有限公司;SHISEIDO CAPCELL PAK C18柱(4.6 mm×150mm,5µm)购于资生堂中国投资有限公司;KQ-500DE型数控超声波清洗器购于昆山市超声仪器有限公司;智能恒温水浴锅购于北京市长风仪器仪表公司;NewClassic MF分析天平购于瑞士梅特勒公司。
二氧化碳(纯度≥99.99 %)购自北京北氧利来公司;甲醇、异丙醇、乙腈、乙醇、甲酸、乙酸、磷酸均为色谱纯,购自Thermo Fisher公司;实验用水来自Milli-Q水纯化系统(电阻率为18.2 MΩ·cm)。对照品:毛蕊异黄酮(M-021-170926)、芒柄花素(C-018-171217)、山奈酚(S-014-160708)、槲皮素(H-009-180615)、异鼠李素(Y-039-160601)、毛蕊异黄酮葡萄糖苷(M-020-170926)、紫云英苷(Z-020-181205)、异槲皮苷(Y-076-161216)、芒柄花苷(M-013-170926),纯度均>98.5 %,购自成都瑞芬思生物科技有限公司。
黄芪药材购自北京同仁堂股份有限公司,留存于北京中医药大学科研楼中药分析与转化研究中心实验室。
1.2 实验方法
1.2.1 供试品溶液的制备 取黄芪药材粉末(过5号筛)约2.00 g,精密称定,置于50mL具塞锥形瓶中,加入30mL甲醇,超声处理30min,重复提取2次,合并提取液,于45℃下蒸发仪浓缩至近干,所得浸膏用甲醇溶解,定容至5mL,过0.45 µm微孔滤膜后,备用。
1.2.2 混合对照品溶液的制备精密称取毛蕊异黄酮、芒柄花素、山奈酚、槲皮素、异鼠李素、毛蕊异黄酮葡萄糖苷、紫云英苷、异槲皮苷、芒柄花苷适量,配成质量浓度分别为4.56 、4.01 、5.25 、6.74 、5.38 、6.08 、5.26 、5.84 、5.43 mg/mL的对照品溶液,于4℃保存备用。
1.2.3 色谱条件 SFC条件为:Agilent ZORBAX RX-SIL色谱柱(4.6 mm×150mm,5µm),流动相为CO2(A)-0.1%磷酸甲醇溶液(B),梯度洗脱:0~4min,10%B;4~15min,10%~25%B。背压:10MPa,柱温:35℃,流速:3mL/min,检测波长:260nm,进样体积:5µL。
HPLC条件为:SHISEIDO CAPCELL PAK C18色谱柱(4.6 mm×250mm,5µm),流动相为水(A)-乙腈(B),梯度洗脱:0~10min,10%~20% B;10~15min,20%~25% B;15~25min,25%~28%B;25~40min,28%~30% B;40~60min,30%~40% B。柱温:30℃,流速:1mL/min,进样量:10µL,进样预平衡:20min。
2 结果与讨论
2.1 SFC分离条件的优化
2.1.1 色谱柱 固定其他色谱条件,考察Agilent ZORBAX RX-SIL(4.6 mm×150mm,5µm)和Waters ACQUITY UPC2TMBEH2-EP(3mm×100mm,1.7 µm)两种色谱柱对9种黄酮类化合物的分离效果,两柱的流速分别设为3、1.3 mL/min,分离结果如图1所示。结果表明:BEH2-EP色谱柱对黄酮类化合物的保留更强,导致化合物在特定条件下未能洗脱。可能是由于SFC的流动相中不含水,在SFC中微弱的分子间作用力(如偶极-偶极作用、电荷转移作用)对化合物和固定相间的保留具有较大影响[16]。因此,吡啶基团中氮原子的氢键作用和吡啶基团与黄酮类化合物产生的π-π共轭作用对化合物保留产生了很大的影响。而9种化合物在RX-SIL色谱柱上达到了基线分离,并有满意的峰形和分离度。以logP为参数解释黄酮类化合物的保留顺序与色谱柱之间的关系,表1为9个黄酮类化合物和两种色谱柱的logP值。由图1可见,洗脱顺序以化合物结构差异分为两类:黄酮类(异鼠李素、山奈酚、槲皮素、紫云英苷、异槲皮苷)和异黄酮类(芒柄花素、毛蕊异黄酮、芒柄花苷、毛蕊异黄酮葡萄糖苷),色谱保留均随logP值的减小而增大,其原因可能是由于黄酮类化合物中含有羟基数目多,极性较大,需要较大极性的色谱柱对其进行保留;同时色谱柱的logP值要小于分析物的logP值,才能达到一定的分离度。因此,选择Agilent ZORBAX RX-SIL色谱柱用于SFC分析。

表1 9种黄酮类化合物及色谱柱的logP值Table1 logP of nine flavonoids and two cloumns
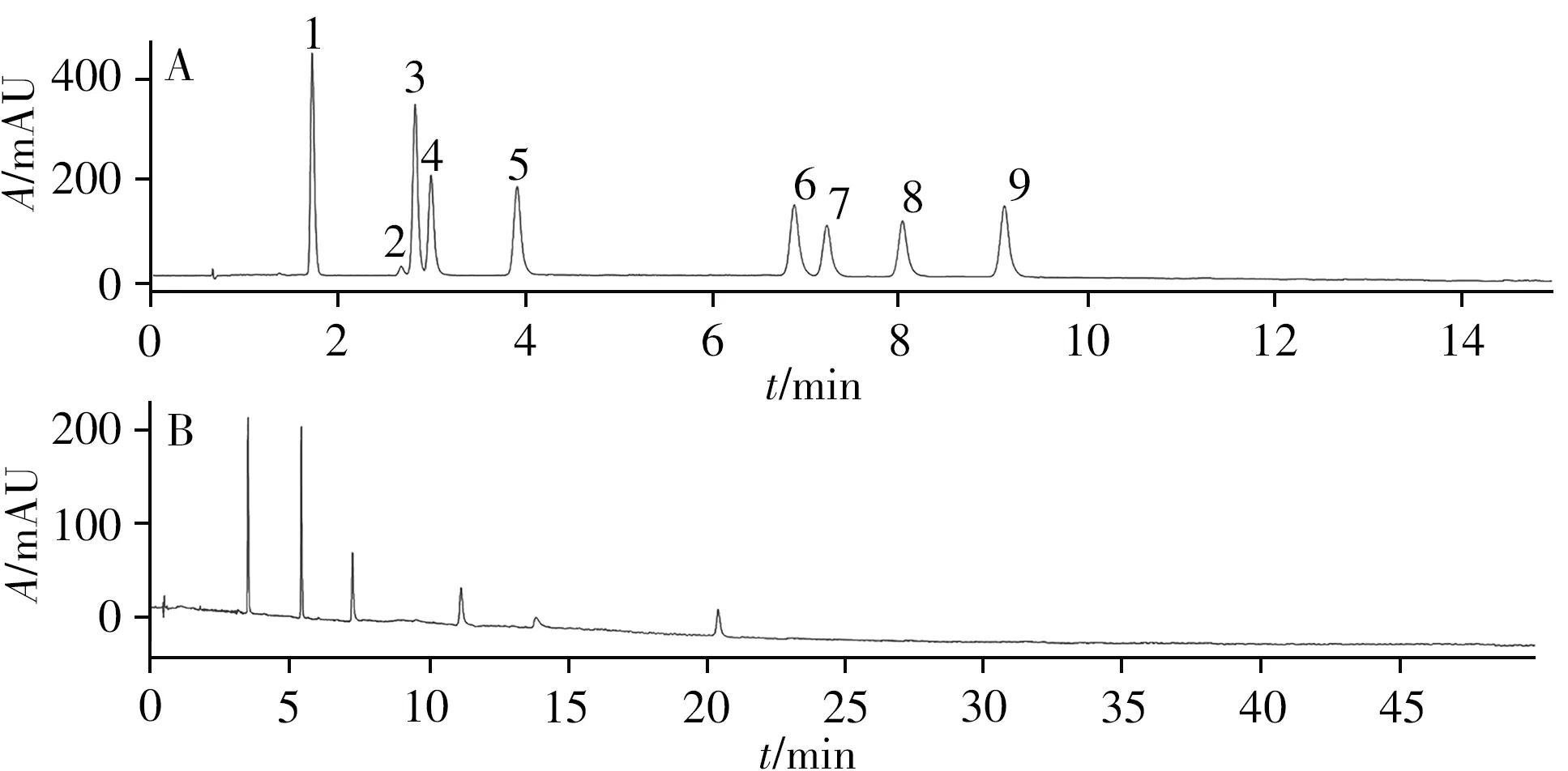
图1 9种黄酮类化合物在两种色谱柱上的分离色谱图Fig.1 SFC chromatograms of nine flavonoids on different columns
2.1.2 改性剂由于CO2极性较弱,需加入改性剂增加超临界CO2流体的极性,从而增加流动相的洗脱和溶解能力。保持其它条件不变,考察了4种不同极性的常用改性剂(甲醇、乙醇、乙腈、异丙醇)对黄芪中9种黄酮类化合物的保留行为,其中乙腈是不具有氢键供体性质的有机溶剂,其他醇类为具有氢键供体性质的有机溶剂[17-18]。结果表明,4种改性剂对化合物的洗脱能力强弱顺序为:甲醇>乙醇>异丙醇>乙腈,当改性剂极性增大时(即流动相极性增大时),黄酮类化合物的溶剂化能力增强,分离效果越好。乙腈对化合物的保留最强,乙醇和异丙醇则会导致不同程度的化合物共洗脱现象。因此,选择甲醇为最佳改性剂。
2.1.3 添加剂由于黄酮类化合物一般含有较多羟基,易与硅胶色谱柱中的硅醇基形成氢键,从而导致黄酮类化合物的色谱峰出现拖尾。为改善这一现象,需在改性剂中加入添加剂(酸、碱或盐)。考察了改性剂中分别添加0.1 %的甲酸、乙酸和磷酸时对9种黄酮类化合物峰拖尾现象的改善程度。结果显示,当添加剂为0.1%磷酸时,9种黄酮类化合物的拖尾因子和峰宽值最小,改善效果最佳。当添加剂为0.1 %甲酸和0.1 %乙酸时,二者虽能竞争抑制黄酮酚羟基与硅醇基形成氢键,对拖尾有一定的改善作用,但由于添加剂在流动相中所占比例很小,因此需加入酸性较强的磷酸来抑制黄酮酚羟基与硅醇基形成氢键。
2.1.4 流速超临界流体的粘度较低,其压力较低,因此可改变的流速范围较广。保持其他色谱条件一致,考察了不同流速下9种黄酮类化合物的色谱保留行为(图2)。结果表明,随着流速的增加,各化合物色谱峰的保留逐渐减小(图2A),且峰形逐渐尖锐(图2B),但由于出峰时间快而降低了部分化合物(异鼠李素和紫云英苷)的分离度,当流速增长至4mL/min时,异鼠李素不能得到有效的洗脱。为保证足够的分离度和较短的分离时间,最终选择流速为3mL/min。
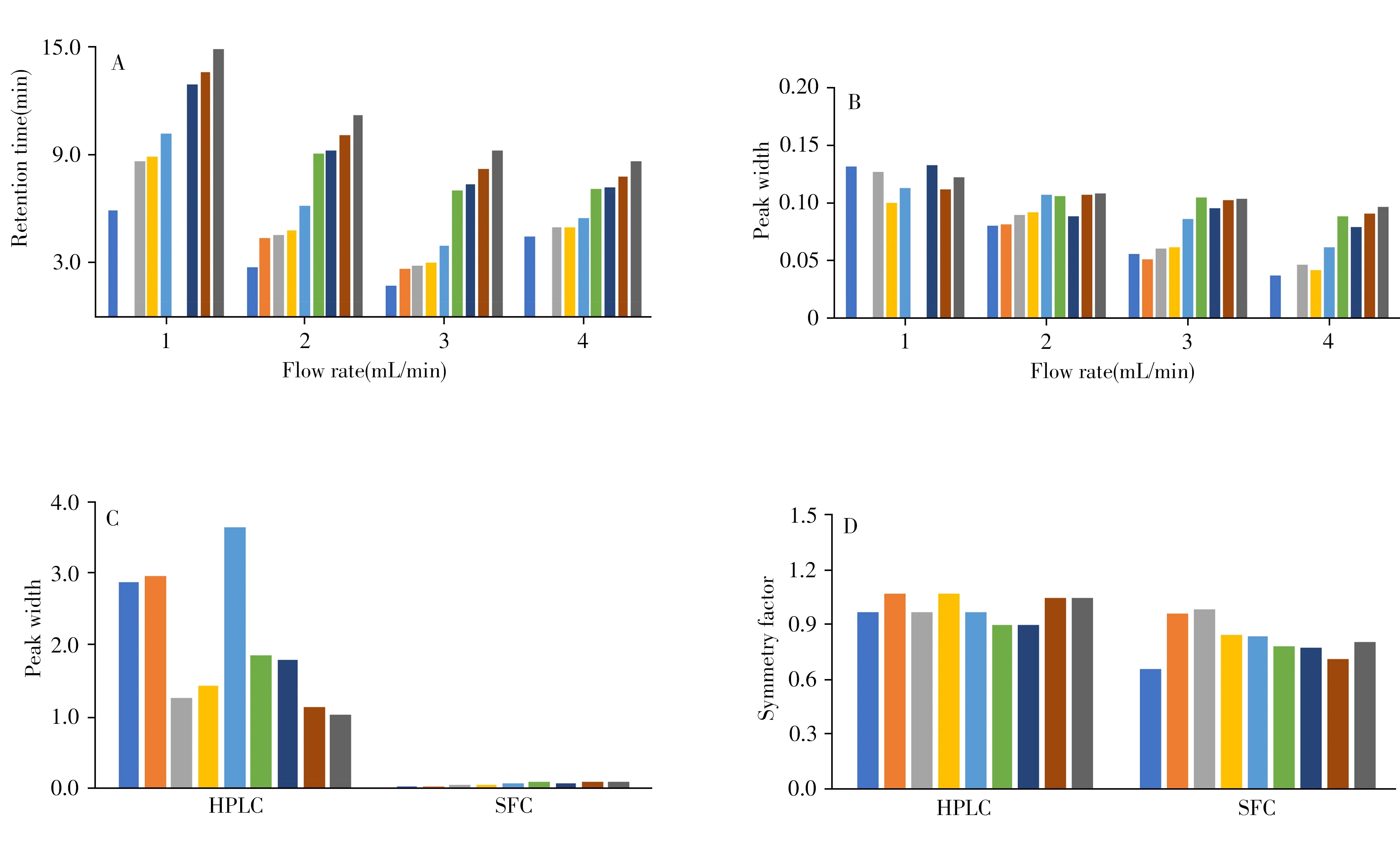
图2 流速对9种黄酮类化合物保留时间(A)及峰宽(B)的影响,以及HPLC法和SFC法对9种黄酮类化合物峰宽(C)和对称因子(D)的影响Fig.2 Effects of flow rate on retention times(A)and peak widths(B)of nine flavonoids,effects of HPLC and SFC methods on peak widths(C)and symmetry factors(D)of nine flavonoids
2.1.5 柱温及背压从流体密度的角度考虑,在SFC中温度和背压通常对分离结果有显著影响。保持其他条件不变,分别考察了不同柱温(30~50℃)和背压(10~15MPa)下9种黄酮类化合物混合标准溶液的分离。结果显示,柱温和背压对黄酮类化合物的分离度影响较小,主要影响化合物的保留值。当温度升高时,超临界CO2流体的密度降低,流动相的溶剂化能力和洗脱能力减弱,导致黄酮类化合物的保留增强;当背压升高时,超临界CO2流体的密度增加,流动相的溶剂化能力和洗脱能力增强,从而使黄酮类化合物的保留减弱。表明当流动相中含有添加剂时,超临界流体状态与亚临界流体是互通的,此时温度和背压可以保持恒定[19]。本实验设定流速为3mL/min,背压为10MPa。
2.2 SFC法与HPLC法分离结果的比较
由“2.1.1 ”可知,固定相的选择对化合物保留至关重要。为深入研究9种黄酮类化合物在不同固定相和不同系统中选择性的区别,比较了9种黄酮类化合物在SFC和HPLC两种系统中的选择性差异。SFC和HPLC的条件如“1.2.3”所示,分离结果如图1A及图3所示。可见,采用SFC法分离9种黄酮类化合物较HPLC法时间缩短了6倍,在SFC法中9种黄酮类化合物于10min内达到基线分离,而在HPLC法中需60min;同时SFC法的色谱峰峰宽均小于0.12,而HPLC法的峰宽均大于1,说明超临界流体与液体相比具有较低的粘度;两种分离方法的对称因子几乎相同,因此具有同样的分离效率(如图2C、D所示)。此外,在HPLC中,毛蕊异黄酮和槲皮素、山奈酚和异鼠李素的分离度小于1,在SFC中这两组化合物可实现基线分离;在SFC中,异鼠李素和毛蕊异黄酮及山奈酚的分离度小于1,而在HPLC中的分离较好。因此SFC和HPLC两种色谱法具有互补性。
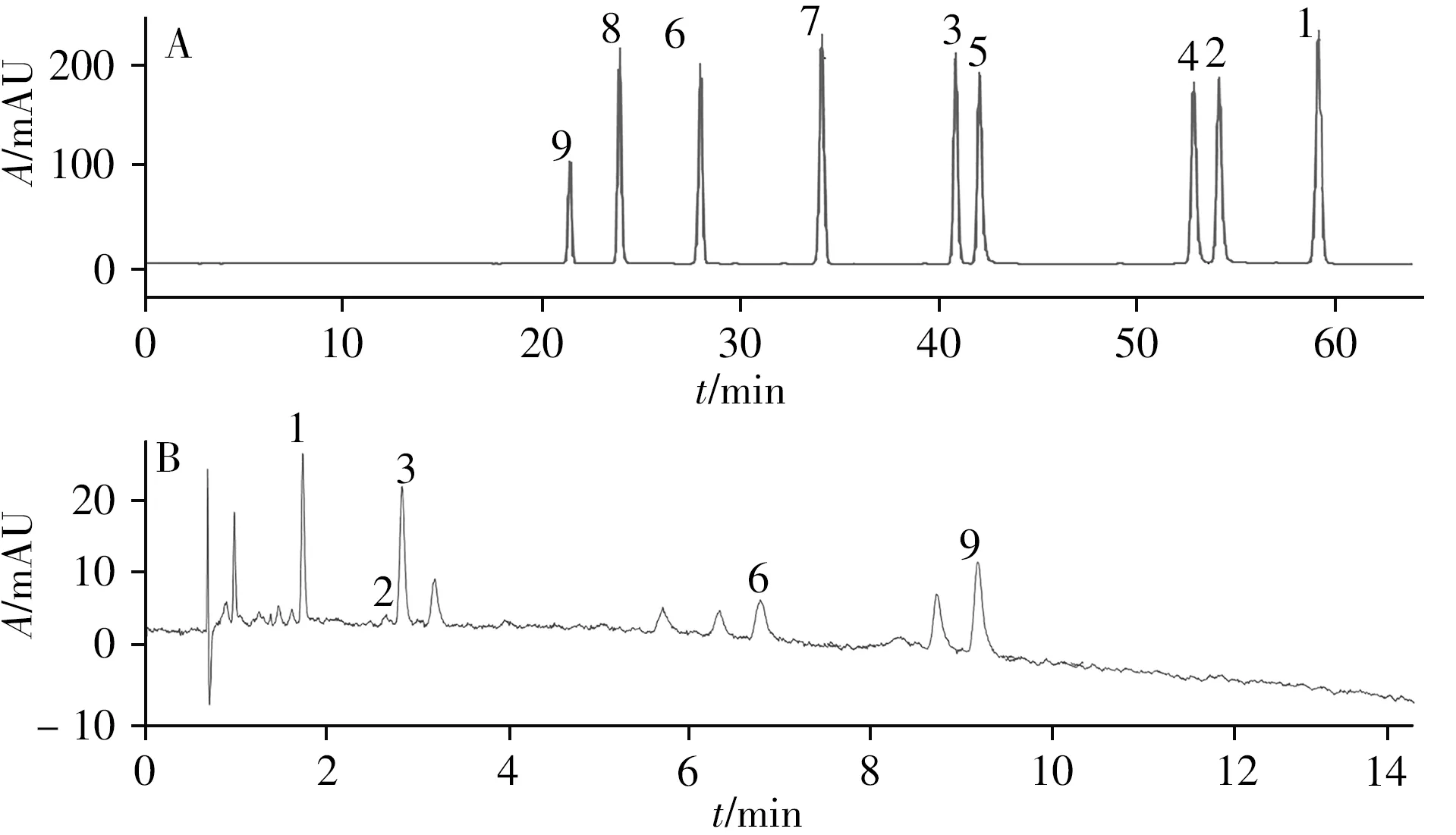
图3 9种黄酮类化合物混合对照品的HPLC图(A)及供试品的SFC图(B)Fig.3 HPLC(A)and SFC(B)chromatograms of nine flavonoids mixed reference substances peak numbers denoted were the same as those in Fig.1
一般认为SFC法的分离机理与正相色谱相似,因此,在理论上SFC和HPLC的保留时间顺序相反。然而根据结果可知:在HPLC系统中,对于糖苷类化合物,毛蕊异黄酮葡萄糖苷、异槲皮苷、芒柄花苷、紫云英苷依次从固定相上洗脱下来;在SFC中,上述4种化合物以相反的保留时间顺序洗脱;对于苷元类化合物,根据其母核可分为异黄酮类(毛蕊异黄酮、芒柄花素)和黄酮类化合物(槲皮素、山奈酚、异鼠李素),在HPLC和SFC系统中,这两类化合物的保留时间相反。上述结果表明,在一定程度上,SFC法与HPLC法具有相反的色谱行为。
2.3 SFC法定量分析
黄芪药材来源广泛,前期研究发现不同黄芪样品中含有的活性成分及其含量存在不一致,如黎映琼等[20]研究表明,芒柄花苷在黄芪根中含量均较叶、茎低,且不同产地的含量差异较大;张妍等[10]研究表明,不同产地黄芪中黄酮类化合物存在较大差异,槲皮素和山奈酚的含量较低;蒲清荣等[21]研究发现,不同产地黄芪中异槲皮苷和芒柄花苷的含量分别为0.0132 ~0.0808 mg/g和0.0021 ~0.0559 mg/g。本研究采用SFC法对黄芪饮片样品进行检测,发现黄芪饮片样品中只检出5种黄酮类化合物(芒柄花素、异鼠李素、毛蕊异黄酮、紫云英苷、毛蕊异黄酮葡萄糖苷)。因此,进一步对上述5种黄酮类化合物进行方法学验证。
2.3.1 线性关系与定量下限 取“1.2.2 ”配制的对照品储备液,按照1∶2.5 ∶5∶2∶2.5 比例稀释,得到6个系列的混合对照品溶液,按照“1.2.3 ”SFC条件进行分析,以黄酮类化合物的质量浓度为横坐标(X,µg/mL),峰面积为纵坐标(Y)绘制标准曲线,计算回归方程及相关系数(r)。以信噪比(S/N)为10时对应的质量浓度作为定量下限(LOQ)。由表2可知,芒柄花素、异鼠李素、毛蕊异黄酮、紫云英苷、毛蕊异黄酮葡萄糖苷在一定的质量浓度范围内线性关系良好,r≥0.9632 ;LOQ为10.69 ~16.21 µg/mL。
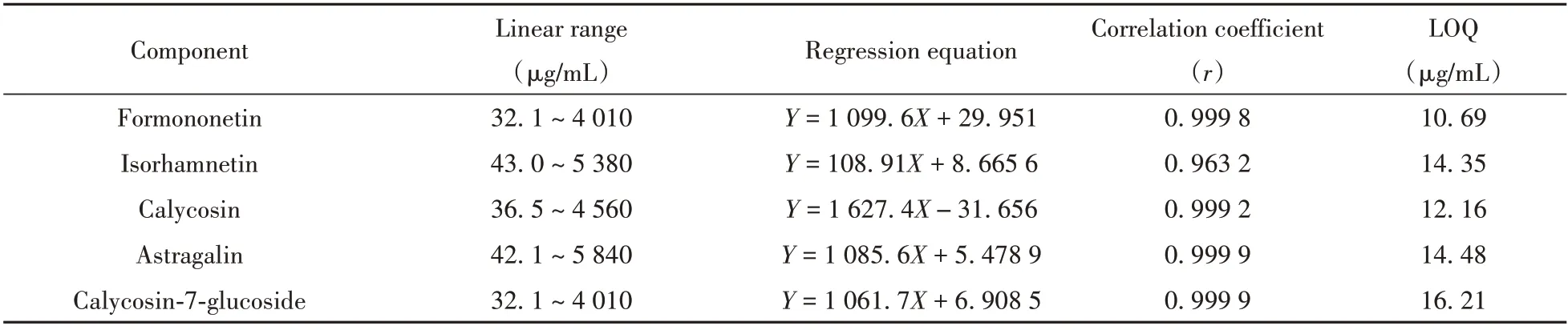
表2 5种黄酮类化合物的线性关系及定量下限Table2 Linear relationships and LOQs of5flavonoids
2.3.2 精密度将5种黄酮类化合物的对照品溶液,按照“1.2.3 ”SFC条件重复进样6次,计算日内和日间相对标准偏差(RSD),以评价日内精密度和日间精密度。5种黄酮类化合物的日内RSD为1.3 %~2.0 %;日间RSD为1.6 %~2.2 %。结果表明,本方法具有较好的日间和日内精密度,可满足黄芪药材中5种黄酮类物质的检测要求。
2.3.3 重复性 精密称取待测的黄芪样品6份,按“1.2.1 ”方法制备供试品溶液,在“1.2.3 ”SFC条件下进样测定并计算含量。结果显示,芒柄花素、异鼠李素、毛蕊异黄酮、紫云英苷和毛蕊异黄酮葡萄糖苷的相对标准偏差(RSD)分别为3.9 %、3.6 %、5.9 %、6.4 %、6.0 %。
2.3.4 稳定性 取黄芪药材粉末,按照“1.2.1 ”方法处理样品,分别于样品制备后1、4、8、12、24、48h,按照“1.2.3”SFC条件测定。黄芪药材中芒柄花素、异鼠李素、毛蕊异黄酮、紫云英苷、毛蕊异黄酮葡萄糖苷的峰面积RSD分别为1.6 %、2.5 %、2.9 %、3.1 %和3.8 %,表明供试品溶液在48h内稳定。
2.3.5 加标回收率 取黄芪药材粉末2.0 g,共9份,分别加入对照品溶液46、92、184µL,配成低、中、高3个加标浓度,每个浓度平行3份,按“1.2.1”方法进行样品制备并检测。计算得到黄芪药材中5种黄酮类化合物的回收率为91.8 %~112%。
2.3.6 样品含量测定 取黄芪药材粉末2.0 g,按照“1.2.1 ”制备供试品溶液,在“1.2.3 ”SFC条件下进行测定。结果表明,芒柄花素、异鼠李素、毛蕊异黄酮、紫云英苷、毛蕊异黄酮苷的含量分别为0.14 、0.024 、0.11 、0.10 、0.09 µg/mg。
根据2020版中国药典规定,黄芪饮片中毛蕊异黄酮葡萄糖苷的含量不少于0.020 %,换算成质量分数即不少于0.2 mg/g[1]。我国黄芪主产区主要包括甘肃、山西、内蒙古3个产地,根据以往研究报道,不同产区的黄芪饮片质量存在差异[22],蒲清荣等[21]采用HPLC法测得不同产地黄芪中毛蕊异黄酮葡萄糖苷的含量为0.0216 ~0.0238 mg/g;李紫岩等[23]测定内蒙古不同产地黄芪中毛蕊异黄酮葡萄糖苷的含量为0.0220 ~0.1454 mg/g;刘亚令等[24]测得不同产地黄芪中毛蕊异黄酮葡萄糖苷的含量为0.004 ~0.034 mg/g。本研究黄芪中的毛蕊异黄酮葡糖糖苷低于中国药典规定的含量范围,主要是产地差异导致。此外,本文建立的SFC法与传统的HPLC法相比,分析时间至少缩短了3倍[7-10],与TLC法[6]相比更准确便捷,证明了这种绿色环保的分析技术在天然药物测定中的应用价值和实际意义。
3 结 论
本文建立了一种可在10min内快速分析9种黄酮类化合物的SFC方法,讨论了色谱柱、改性剂、添加剂、流速、柱温、背压等因素的影响,比较了SFC和HPLC两种色谱模式的选择性差异,并对黄芪中5种黄酮类化合物含量进行测定。虽然建立SFC法时影响化合物分离的因素较多,但研究发现化合物的分离效果受色谱柱影响显著,其保留及选择性受改性剂和添加剂影响大,且添加剂对色谱峰形的影响明显。此外,SFC和HPLC这两种色谱方法具有互补性,在一定程度上,其色谱保留行为相反。虽然由于SFC分离机制的不确定性和复杂性,导致无法快速筛选色谱柱,且存在仪器稳定性差、对样品的前处理要求相对较高等缺点。然而作为中药中的一种新型色谱技术,SFC仍存在值得挖掘的价值。
猜你喜欢
城市道桥与防洪(2022年7期)2022-08-31
食品工业(2022年5期)2022-06-13
中国药房(2022年10期)2022-05-30
食品与健康(2020年4期)2020-05-18
筑路机械与施工机械化(2019年5期)2019-11-30
筑路机械与施工机械化(2019年10期)2019-11-05
分析化学(2018年4期)2018-11-02
中国食品(2018年7期)2018-09-10
成长·读写月刊(2017年7期)2017-07-13
家庭医药·快乐养生(2017年4期)2017-04-19
