
快速固相萃取/超高效液相色谱-串联质谱法测定水产品中25种磺酰脲类及磺酰胺类除草剂残留
2021-10-17 07:58朱富强吴树栋韩岩君郭宇鹏
分析测试学报 2021年9期
朱富强,吴树栋,韩岩君,郭宇鹏
(滨州市检验检测中心,山东 滨州 256600)
磺酰脲类和磺酰胺类除草剂在世界农药市场中占有重要地位,两者应用范围广、用量大,对于水稻、玉米、大豆、小麦、甜菜、油菜等作物的杂草防治具有良好效果[1-3]。磺酰脲类除草剂和磺酰胺类除草剂在施用过程中易通过地表径流和渗透进入水环境中,污染养殖水体,进而影响水体生物的生长发育,造成水产品质量安全隐患,危害消费者健康。我国农业部公告第2032号[4]规定,于2015年12月31日起全面禁止使用氯磺隆,于2017年7月1日起全面禁止使用胺苯磺隆及甲磺隆,并在GB2763-2019[5]中规定了多种磺酰脲类及磺酰胺类除草剂在食品中的最大残留限量,但尚未制定在动物源性食品中的最大残留限量。日本肯定列表制度中规定了动物源性食品中磺酰脲类及磺酰胺类除草剂的最大残留限量[6],如噻吩磺隆及苯磺隆在猪肉等肉类中的最大残留限量为0.01 mg/kg,磺酰磺隆在鱼类、软体动物、甲壳纲动物中的最大残留限量为0.005 mg/kg,唑嘧磺草胺在鸡肉、肝脏、蛋和奶中的最大残留限量为0.1 mg/kg。因此,建立水产品中磺酰脲类及磺酰胺类除草剂的检测方法,对于保障进出口贸易、保护人民健康具有重要意义。
目前,文献报道的水产品等动物源性食品中农药残留的检测方法主要有气相色谱法(GC)[7-8]、气相色谱-质谱联用法(GC-MS)[9-10]、气相色谱-串联质谱法(GC-MS/MS)[11-12]、液相色谱法(HPLC)[13-14]及液相色谱-串联质谱法(LC-MS/MS)[15-16]等,样品净化方式通常采用固相萃取法(SPE)[17-18]和QuEChERS法等[19-20]。传统的SPE方法操作费时、步骤繁琐,影响结果稳定性和检验效率,不能满足大批量样品的高通量、快速检测要求。QuEChERS法具有便捷、高效、适用范围宽等优点,但净化效果往往不如SPE法,且在实际应用过程中需对各吸附净化剂(如N-丙基乙二胺、十八烷基硅烷键合硅胶、石墨化碳黑等)以及盐析剂的用量进行优化[15-16,19-20]。SHIMSEN QVet-NM+固相萃取柱使用新一代的固相萃取吸附剂,无需活化、淋洗、洗脱,可直接将提取液上样在正压下过滤除去杂质,实现通过式一步净化,过程简便、高效。本文针对不同基质类型的水产品,采用SHIMSEN QVet-NM+固相萃取柱净化,以超高效液相色谱-串联质谱(UPLC-MS/MS)法测定样品中25种磺酰脲及磺酰胺类农药残留。该方法净化效率高、操作便捷,且灵敏度高、准确性好,可为水产品中农药残留风险监控提供借鉴与参考。
1 实验部分
1.1 仪器与试剂
ACQUITY UPLC I-Class超高效液相色谱仪、Xevo TQ-XS三重四极杆质谱仪(美国Waters公司);AccucoreTMaQ C18色谱柱(2.1 mm×50mm×2.6 µm,美国Thermo Fisher公司);Shim-pack GIST C18色谱柱、MultiS-100涡旋混合器(岛津(上海)实验器材有限公司);3-18K5离心机(德国Sigma公司);CP225D电子分析天平(德国Sartorius公司);Milli-Q Advantage A10超纯水仪(美国Millipore公司)。
甲醇、乙腈(HPLC级,德国Merck公司);甲酸(纯度>99%,美国ACS恩科化学公司);超纯水(电阻率≥18.2 MΩ∙cm);SHIMSEN QVet-NM+固相萃取柱(5mL/5g)、SHIMSEN QuEChERS提取盐包(含1g氯化钠、4g硫酸镁、0.5 g柠檬酸二钠盐水合物、1g无水柠檬酸钠,岛津(上海)实验器材有限公司);尼龙针式滤器(0.22 µm,上海安谱科技有限公司)。
标准品:醚磺隆(100µg/mL)、甲酰胺磺隆(100µg/mL)、氯酯磺草胺(100µg/mL)、玉嘧磺隆(1000µg/mL)购自AccuStandard公司;甲基噻吩磺隆、吡嘧磺隆的质量浓度均为100µg/mL,购自ANPEL公司;氟唑嘧磺草胺的质量浓度为100µg/mL,购自BePure公司;氟磺隆、氟嘧磺隆、氟胺磺隆、五氟磺草胺、双氯磺草胺、醚苯磺隆、甲磺隆、双氟磺草胺、氯磺隆、四唑嘧磺隆、环丙嘧磺隆的质量浓度均为100µg/mL,购自First Standard公司;苯磺隆(100µg/mL)购自农业农村部环境保护科研监测所;氯嘧磺隆的质量浓度为100µg/mL,购自北京坛墨质检科技有限公司;咪唑磺隆、嘧啶磺隆、磺酰磺隆、乙氧嘧磺隆、胺苯磺隆的纯度均≥99%,购自Dr.Ehrenstorfer公司,各准确称取10.0 mg,用甲醇稀释定容至100mL容量瓶中,配成质量浓度均为100µg/mL的标准储备液,于-20℃避光保存。
1.2 标准溶液的配制
各准确移取一定量25种待测化合物标准溶液至50mL容量瓶中,用乙腈稀释定容至刻度,配制成1µg/mL的混合标准中间液,于-20℃低温保存。临用前,用空白样品提取液配制成系列质量浓度的基质混合标准工作溶液。
1.3 样品前处理
分别取草鱼、鲤鱼、鲫鱼、克氏原螯虾、南美白对虾、文蛤的可食部分进行均质,准确称取2g试样(精确至0.01 g)至50mL离心管中,加入10.0 mL超纯水,涡旋1min,再准确加入10.0 mL乙腈,涡旋提取5min,然后加入SHIMSEN QuEChERS提取盐包,剧烈振摇1min,以8000r/min离心5min。取5mL上清液通过SHIMSEN QVet-NM+固相萃取柱,以1滴/s的流速加压萃取,收集滤液,准确吸取500µL滤液与500µL超纯水混合,过0.22 µm有机微孔滤膜后,待测。
1.4 色谱与质谱条件
色谱条件:Shim-pack GIST C18色谱柱(2.1 mm×50mm×2.0 µm);柱温为40℃;进样体积为5µL;流速为0.40 mL/min;流动相:A为0.1 %甲酸水溶液,B为乙腈。梯度洗脱程序为:0~2.8 min,10%~70% B;2.8 ~2.9 min,70%~95% B;2.9 ~3.9 min,95% B;3.9 ~4.0 min,95%~10% B;4.0 ~6.0 min,10%B。
质谱条件:电喷雾电离(ESI)源,正、负离子模式同时扫描;多反应监测模式(MRM);离子源温度:150℃;毛细管电压:2.00 kV;脱溶剂气温度:600℃;脱溶剂气流量:1000L/h;锥孔反吹气流量:150L/h;碰撞气流量:0.15 mL/min。25种除草剂的质谱分析参数见表1。
2 结果与讨论
2.1 仪器条件的选择
2.1.1 质谱条件的优化 以0.1 %甲酸水溶液-乙腈(30∶70)为流动相,采用流动注射的方式,对待测化合物的质谱条件进行优化。在电喷雾正、负离子同时监测模式下,优化离子源温度、脱溶剂气温度、毛细管电压、锥孔电压等参数,使目标物母离子的响应信号强度达到最佳,并进行二级质谱扫描,确定定性离子和定量离子,在多反应监测模式下优化碰撞能量等质谱参数。优化后的质谱参数见表1。
2.1.2 色谱条件的优化 分别考察了AccucoreTM aQ C18色谱柱(2.1 mm×50mm×2.6 µm)和Shimpack GIST C18色谱柱(2.1 mm×50mm×2.0 µm)对25种待测化合物的分离效果,结果发现采用后者作为分离柱时各待测化合物的峰响应值更高,且保留时间更为合理,故选用Shim-pack GIST C18色谱柱为分离柱。由于待测物大部分在正离子模式下测定,酸性条件有利于离子化,可增强其响应值,故流动相A相采用0.1%甲酸水溶液。比较了乙腈、甲醇作为流动相B相对目标物色谱分离行为的影响。结果表明,以甲醇作为流动相B相时,25种待测化合物的峰响应值普遍较低,且乙氧嘧磺隆、醚磺隆的峰形拖尾明显;以乙腈作为流动相B相时,25种待测化合物的峰响应值均高于甲醇,且峰形尖锐,因此采用0.1%甲酸水溶液-乙腈作为流动相。MRM模式下25种待测化合物混合标准溶液(10µg/L)的总离子流图(TIC)见图1。

图1 MRM模式下25种待测化合物混合标准溶液(10µg/L)的总离子流图Fig.1 Total ion chromatogram of25analytes mixed standard solution(10µg/L)in MRM mode
2.2 样品提取方法的优化
本实验尝试采用甲醇、乙腈作为提取溶剂,结果显示,由于水产品为高蛋白样品,直接加入强极性有机溶剂后使蛋白质迅速失水变性,导致样品迅速结块,影响待测化合物的提取效率。在加入提取溶剂前,加水稀释使样品分散均匀有利于待测化合物的提取。
进一步以阴性草鱼样品为实验对象,在待测化合物加标水平为10µg/kg条件下,首先加入10mL水使样品分散,再分别加入10mL的不同提取溶剂(甲醇、1%甲酸甲醇、乙腈及1%甲酸乙腈),然后加入QuEChERS提取盐包进行盐析,考察各提取溶剂对待测化合物的提取效率。结果表明,经甲醇或1%甲酸甲醇提取后,盐析效果差,无法分层,且所得提取液较为浑浊,待测化合物的回收率普遍较低(0%~45.6%),其原因可能在于甲醇水溶液对干扰物的溶解能力强,共萃物较多,影响了待测化合物的测定。采用乙腈或1%甲酸乙腈作为提取溶剂时各待测化合物的回收率均大于62%,且两者无明显差异。为节约成本、安全环保,最终选用乙腈作为提取溶剂。
2.3 样品净化方式的选择
经乙腈提取后的样品溶液中含有大量磷脂等脂溶性化合物,会干扰待测化合物测定,且影响仪器性能。为降低基质干扰,提高方法的准确性和灵敏度,减少对色谱柱、质谱检测器的污染,须对样品进行有效的净化处理。SHIMSEN QVet固相萃取柱包括QVet-AG+、QVet-HP+、QVet-NM+等型号,前两者主要应用于高极性化合物,后者的适用范围宽,可应用于不同类型化合物的净化。考虑到待测化合物的理化性质,因此选择SHIMSEN QVet-NM+快速固相萃取柱对提取液进行净化。采用通过式固相萃取净化策略,将5mL样品提取溶液直接加载至固相萃取柱以除去磷脂等杂质,过程简便、快速、高效。为考察其净化效果,参考文献[21]的方法,通过采集m/z184前体离子,得到磷脂类化合物的含量信息。图2为经SHIMSEN QVet-NM+固相萃取柱净化前后空白草鱼样品溶液的m/z184的TIC图,可见,经净化后有效去除了磷脂类化合物,表明该方法的净化效果显著。
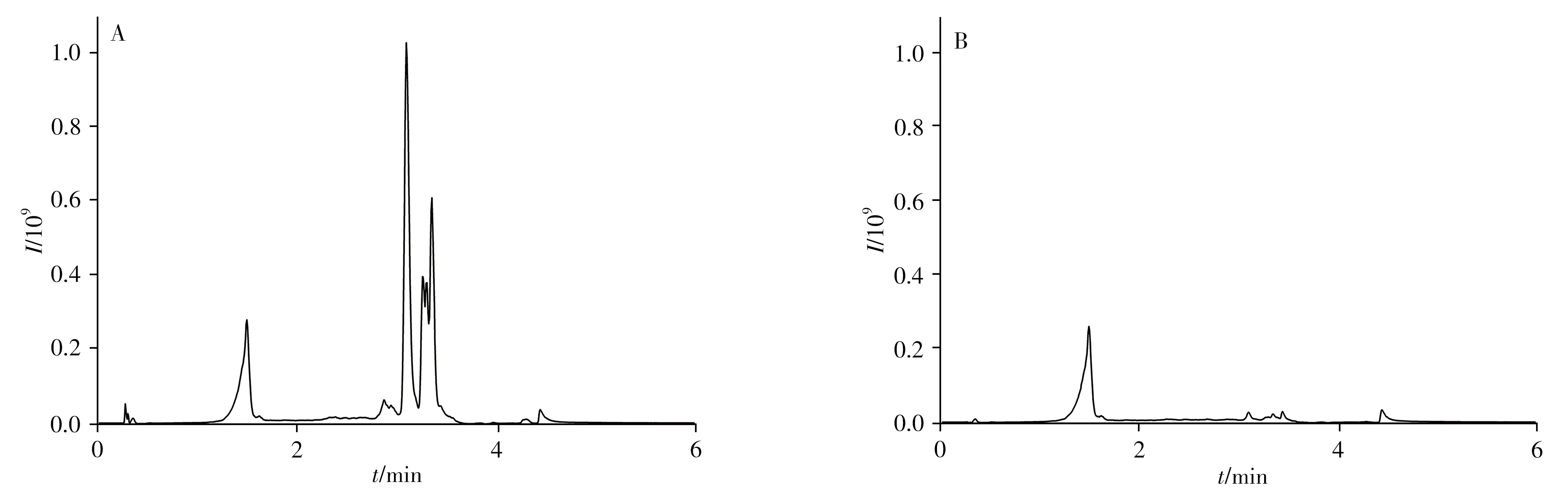
图2 采用快速固相萃取柱净化前(A)和净化后(B)的空白草鱼样品检测效果比较Fig.2 Comparison of blank grass carp samples before(A)and after(B)rapid solid phase extraction
2.4 进样溶剂的优化
净化后所得滤液的溶剂为乙腈,将滤液直接上机测试,发现所有待测化合物的峰形较宽、峰前拖尾,峰面积重现性较差,其原因为样品溶剂的洗脱强度大于流动相的洗脱强度,溶剂效应导致峰变形及柱效降低。本文采用稀释法减小溶剂效应,经试验发现,取500µL滤液与500µL超纯水混合后进样分析,所有待测化合物的峰形均正常,且不会对检出限造成较大影响,故最终采用将滤液用超纯水稀释1倍后进样分析。
2.5 样品基质效应的考察
在LC-MS/MS分析中,基质效应(ME)是影响定性定量结果准确性的重要因素之一。计算公式为:ME=(基质匹配校准曲线斜率/溶剂配制校准曲线斜率-1)×100%[22]。|ME|<20%为弱基质效应,20%≤|ME|≤50%为中等程度基质效应,|ME|>50%为强基质效应。由表2可知,25种待测化合物在草鱼、南美白对虾、文蛤中均表现为基质抑制效应(ME<0);在草鱼基质中,玉嘧磺隆、氯磺隆、醚苯磺隆、氟磺隆表现为中等程度基质效应;在南美白对虾基质中,苯磺隆、玉嘧磺隆、磺酰磺隆、甲磺隆、醚苯磺隆、氟磺隆表现为中等程度基质效应;在文蛤基质中,氟唑嘧磺草胺、甲基噻吩磺隆、苯磺隆、醚磺隆、磺酰磺隆表现为中等程度基质效应。其余待测化合物在草鱼、南美白对虾、文蛤基质中均为弱基质效应。为确保结果准确性,选择基质匹配标准溶液外标法进行定量分析。

表2 25种待测化合物在草鱼、南美白对虾及文蛤中的基质效应Table2 Matrix effects(ME)of25analytes in grass carp,penaeus vannamei and meretrix meretrix
2.6 线性关系与检出限
分别采用草鱼、南美白对虾、文蛤空白基质溶液配制质量浓度为0.20 ~50µg/L的混合标准工作溶液,按照前述的仪器条件进行测定,以待测化合物的质量浓度为横坐标(X,µg/L),峰面积响应值为纵坐标(Y)建立标准曲线。并以不同加标水平下空白基质样品的仪器响应为基准,按信噪比S/N=3计算检出限(LOD),S/N=10计算定量下限(LOQ)。在3种空白样品中,25种待测化合物在0.20 ~50µg/L范围内呈良好的线性关系,相关系数(r2)均大于0.994 ;LOD为0.30 ~1.3 µg/kg,LOQ为1.2 ~5.0 µg/kg。以草鱼空白基质为例,结果见表3。

表3 草鱼空白基质中25种待测化合物的线性方程、相关系数、检出限、定量下限、回收率及相对标准偏差(n=6)Table3 Linear equations,correlation coefficients(r2),LODs,LOQs,recoveries and RSDs(n=6)of25analytes in blank grass carp
2.7 加标回收率与相对标准偏差
取草鱼、南美白对虾、文蛤空白基质样品,进行低、中、高3个浓度水平的加标回收实验,按上述实验条件进行测定,每个加标水平平行分析6次,计算各目标物的回收率和相对标准偏差(RSD)。结果表明,该方法在草鱼空白基质中3个加标水平下的回收率为62.7 %~117%,RSD为1.5 %~18%;在南美白对虾空白基质中的回收率为66.1 %~122%,RSD为1.8 %~17%;在文蛤空白基质中的回收率为76.4 %~115%,RSD为0.72 %~11%。以草鱼为例,各待测化合物的回收率和RSD见表3。
2.8 实际样品的测定
应用本方法对本实验室在监督抽检中留存的10份水产品(包括草鱼、鲤鱼、鲫鱼、克氏原螯虾、南美白对虾、文蛤)进行了测定,结果显示所有样品均未检出待测化合物。
3 结 论
本文结合快速固相萃取技术,以超高效液相色谱-串联质谱建立了水产品中25种磺酰脲类和磺酰胺类农药残留的分析方法。该方法前处理简便快速、净化效果显著,且灵敏度高、准确性好,能够准确地对水产品中上述25种农药残留进行定性定量分析,为水产品等动物源性食品中农药残留的监测与控制提供了一种新的可行途径。
猜你喜欢
当代水产(2022年7期)2022-09-20
煤化工(2022年3期)2022-07-08
当代水产(2022年5期)2022-06-05
小学阅读指南·低年级版(2022年5期)2022-05-09
当代水产(2022年3期)2022-04-26
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
当代水产(2021年10期)2022-01-12
广东第二课堂·小学(2018年9期)2018-10-24
发明与创新·中学生(2018年10期)2018-10-15
食品界(2017年9期)2017-09-30
