
工业NMP 含量两种分析方法讨论
2021-10-14 09:33叶润华赵佳琪刘宝珠
化学工程师 2021年8期
叶润华,赵佳琪,刘宝珠
(新疆美克化工有限责任公司 化验室,新疆 库尔勒 841000)
在天然气部分氧化制乙炔生产工艺过程中,工业N-甲基-2-吡咯烷酮(俗称NMP)作为溶剂,在工艺提浓工段裂解气和循环气的混合气用NMP 提取乙炔气[1]。作为入厂原料,需要快速准确分析NMP含量判断其是否符合要求指标(一般要求指标≥99.0%),为生产工艺提供参考数据。凯氏定氮法是一种通用国家标准[2],广泛用于各种食品、谷物、饲料等样品的蛋白质含量测定[3],此法分为常量、半微量和微量3 种,整个测定过程分为消解、蒸馏、滴定3 步,最后通过吸收氨的量计算NMP 含量;此方法操作难度大,耗时长,需要近8h 才能得出分析结果。但凯氏法在各种定量法中仍占中心地位,当报道一种新方法时,通常是将新法测定结果与凯氏法相比较,并探讨其相关性[4]。为满足生产分析需求,色谱快速分析方法近年来逐渐替代凯氏定氮法用于分析NMP 含量,其原理主要是用卡尔费休仪分析出样品中微量水含量,用自动电位滴定仪分析出微量总胺含量,再通过差减杂质利用色谱面积归一化法计算出NMP 含量[5],该方法准确、快速,仅需1.5h 即可得出分析结果,与凯氏定氮法相比,省时省力,可快速准确为生产提供分析数据。凯氏定氮法分析结果较气相色谱法略高0.4%左右,但两种分析结果均符合要求指标。下面就两种分析方法进行讨论。
1 凯氏定氮法
1.1 实验原理
在CuSO4、硒粉及K2SO4作用下,有机含氮化合物在热浓H2SO4中进行消解,有机氮被固定为(NH4)2SO4,消解完后加入过量浓碱,蒸馏,释放出的NH3用H3BO3溶液吸收,并用HCl 标准溶液滴定。
1.2 实验试剂
H2SO4(AR 西龙科学股份有限公司);甲基红-溴甲酚绿混合指示剂;40g·L-1H3BO3溶液(AR 国药集团化学试剂有限公司);400g·L-1NaOH 溶液(AR 西龙科学股份有限公司);0.1000mol·L-1HCl 标准滴定溶液(优级纯 国药集团化学试剂有限公司);混合消化剂;硒粉(AR 天津市化学试剂研究生);0.5g CuSO4(AR 天津市永晟精细化工有限公司);1g K2SO4(AR国药集团化学试剂有限公司) 为1∶2,20g 混合后研磨均匀。
1.3 操作步骤
(1)称取试样约0.1g(精确至0.2mg)于预先加有混合消化剂2g 的500mL 凯氏定氮瓶中,沿壁加入20mL 浓H2SO4,按消化要求搭好设备,使溶液温度保持在沸点以下缓慢加热。待泡沫停止发生后,加热使其沸腾,溶液由黑色转为透明,继续加热消化2~3h。
(2)冷却后缓慢加入250mL 蒸馏水,摇匀,冷却。沿壁缓慢加入100mL NaOH(400g·L-1)至瓶底,自成一液层后加入几粒沸珠。
(3)摇匀后按蒸馏要求搭好设备,用加有H3BO350mL 和甲基红-溴甲酚绿指示剂的500mL 三角瓶吸收蒸馏的冷凝液,当蒸馏出2/3 液体至三角瓶后停止加热(蒸馏时间一般控制在40min)。用少量蒸馏水冲洗冷凝管,用0.1000mol·L-1的HCl 标准滴定溶液滴定至终点,同时做空白实验。
(4)结果计算
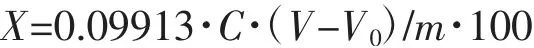
式中 X:NMP 质量分析含量,%;C:HCl 标准滴定溶液浓度,mol·L-1;V:样品吸收液消耗HCl 标准溶液体积,mL;V0:空白样品吸收液消耗HCl 表顺溶液体积,mL;m:样品质量,g。
1.4 影响分析结果的因素
(1)在凯氏定氮操作过程中每个环节都有可能产生误差,若精心操作可将误差减小到最低限度,使测定结果准确可靠。(2)在消解阶段要将试样消解完全,否则分析结果会偏低。(3)在蒸馏阶段,与玻璃器皿相联接的每个接头均需确保密封不漏气,所使用的乳胶管要检查是否老化漏气,否则蒸馏出的NH3不能被完全吸收,会使结果偏低。(4)加入NaOH 溶液时要缓慢加入,防止NH3逃逸使结果偏低。
2 气相色谱法
2.1 实验原理
原料样品工业NMP 中的杂质在载气作用下通过30m 长的(50%-氰丙基苯基)-二甲基聚硅氧烷作固定相的毛细管柱,经与固定相的作用各组分被分离并流出色谱柱,通过FID 检测器检测各组分含量。样品中微量水用卡尔费休仪分析,总胺用自动电位滴定仪分析,NMP 含量用面积归一化法定量计算。
2.2 实验仪器、试剂
7890A 型气相色谱仪(配有FID 检测器、程序升温系统和分流进样器,美国Agilent);30m 毛细管柱,(固定相(50%-氰丙基苯基)-二甲基聚硅氧烷膜);870 型卡尔费休微水分析仪(瑞士万通);916 型自动电位滴定仪(瑞士万通)。
丙醇(优级纯 天津光复精细化工研究所);0.02000mol·L-1的HCl 标准滴定溶液;卡费试剂(无吡啶,3.0mg·mL-1科隆化学品有限公司)。
2.3 气相色谱分析条件
载气:N20.5~1.5mL·min-1;分流比:25∶1;燃气:H2;助燃气:钢瓶空气;进样器温度:250~350℃;柱箱温度,程序升温:初始温度,72~120℃保持2min;加热速率:8~12℃·min-1;最终温度:160~200℃保持15min;检测器温度:FID 检测器250~350℃;进样量:0.2μL。
2.4 实验步骤
2.4.1 样品的总胺含量分析
(1)方法原理 试样用异丙醇溶解,以HCl 标准溶液滴定试样中的碱性物质,用自动电位滴定仪指示滴定终点,总胺含量以一甲胺计算得到。
(2)测定步骤 准确称取试样65g,精确至0.01g,置于250mL 玻璃烧杯中,加入100mL 异丙醇溶液溶解,摇匀后在自动电位滴定仪上用非水相pH 电极滴定至终点。
(3)总胺含量结果计算 以质量百分数表示试样中的总胺含量,公式如下:
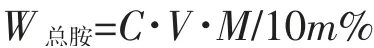
式中 C:HCl 标准滴定溶液浓度,mol·L-1;V:试样消耗HCl 标准滴定溶液体积,mL;M:一甲胺(CH3NH2)摩尔质量,31.06g·mol-1;m:称取试样的质量,g。
2.4.2 样品中微量水含量的分析 使用卡尔费休仪分析试样中微量水含量,用质量百分含量W水表示。
2.4.3 样品的色谱分析
(1)相对校正因子的计算 在分析天平上准确称量100.0000g N-甲基-2-吡咯烷酮,0.1000g γ-丁内酯,0.1000g1,4-丁二醇,0.1000g 2-吡咯烷酮于100mL 容量瓶内,混匀该溶液,在色谱仪上进样0.2μL。
各组分相对校正因子计算公式为:
校正因子=样品百分含量%/面积百分比含量。
(2)色谱进样 在色谱仪上直接进样品0.2μL。
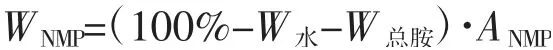
(3)样品中NMP 百分含量的计算式中 WNMP:样品中NMP 的质量百分含量,%;W水:样品中微量水的质量百分含量,%;W总胺:样品中总胺的质量百分含量,%;ANMP:样品中NMP 的面积百分比,%。
(4)样品色谱图见图1。

图1 样品色谱图Fig.1 Chromatogram of sample
3 结果与讨论
3.1 两种方法分析结果对比
选取5 个不同批次的样品分别用凯氏定氮法和气相色谱法分析NMP 含量,以验证气相色谱法的准确性,分析结果见表1。
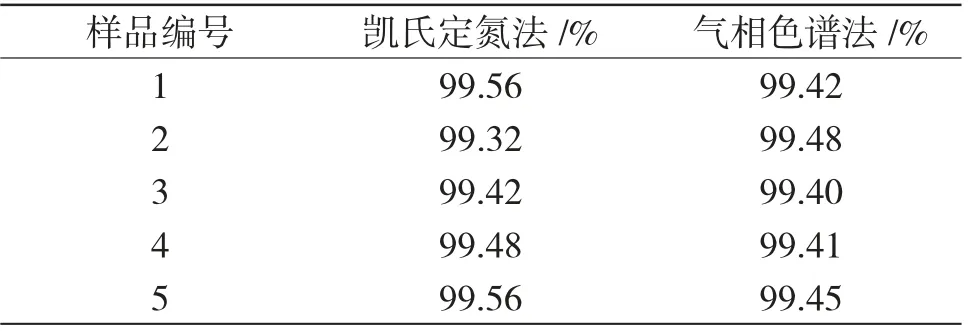
表1 凯氏定氮法与气相色谱法分析结果对比Tab.1 Comparison of analytical results between Kjeldahl method and gas chromatography
由表1 可以看出,同一样品分别用凯氏定氮法和气相色谱法分析NMP 含量,分析结果最大绝对差值在0.2%以内,分析结果均符合要求指标(≥99.0%)。分析同一个样品的NMP 含量,使用凯氏定氮法需8h 左右才能出结果,但所需分析设备简单易得;使用气相色谱法在1.5h 内就可得出分析结果,极大缩短分析时间,且操作简便,省时省力,但需要气相色谱、自动电位滴定仪等设备,两种分析方法各有优点和缺点。但为满足生产及时性需要,目前主要使用气相色谱法。
3.2 气相色谱法精密度实验
用气相色谱法对3 个不同样品分析其NMP 含量,每个样品做5 次平行实验,结果见表2。

表2 气相色谱法精密度实验Tab.2 Precision experiment of gas chromatography
由表2 可以看出,气相色谱法的标准偏差为0.005~0.008 之间,相对标准偏差为0.005%~0.008%之间,方法的精密度较高。
4 结论
(1)工业NMP 含量分析,可以使用凯氏定氮法和气相色谱法两种分析方法,两种方法分析结果绝对差值在0.2%以内,均在要求指标范围内(≥99.0%)。
(2)两种分析方法各有优点和缺点,凯氏定氮法所需分析设备简单易得,但分析操作难度较大,耗时较长,需8h 左右才能出结果;气相色谱法分析操作简便,省时省力,但需要气相色谱仪和自动电位滴定仪等设备。
(3)从满足工业生产及时性需求方面考虑,使用气相色谱法可快速准确得出分析结果。
猜你喜欢
当代水产(2022年4期)2022-06-05
供水技术(2022年1期)2022-04-19
环境保护与循环经济(2021年7期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
陶瓷学报(2021年4期)2021-10-14
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
保鲜与加工(2021年1期)2021-02-06
中国金属通报(2020年4期)2020-12-20
Annals of Applied Mathematics(2020年3期)2020-09-14
