
非典型Stickler综合征I型三个家系的遗传分析及产前诊断
2021-10-09 11:20白周现邵敬芝吴庆华孔祥东
中华眼视光学与视觉科学杂志 2021年9期
白周现 邵敬芝 吴庆华 孔祥东
作者单位:1郑州大学第一附属医院妇产科 遗传与产前诊断中心 450000;2郑州大学第一附属医院眼科 450000
Stickler综合征I型(Stickler syndrome type I,STL1)是一种罕见的常染色体显性遗传胶原结缔组织病,主要以眼部异常、颅颌面畸形、听力损伤及关节异常为特征性病变[1]。其眼部病变尤为突出,表现为高度近视、玻璃体变性、白内障、复发性视网膜脱离等。已知COL2A1基因(Collagen type II alpha 1 chain)OMIM*120140(人类孟德尔遗传在线数据库基因编号)与Stickler综合征相关,COL2A1基因变异可导致STL1 OMIM#108300 或OMIM#609508(即非典型STL1,非综合征性眼病)。非典型STL1患者与STL1患者相比通常仅有严重的眼部表型或伴有身体其他系统轻微表型[2,3]。由于STL1涉及多器官且表型复杂,非典型STL1的确诊较为困难,基因检测对该病的确诊及鉴别具有重要参考意义。本研究收集到3个非典型STL1家系,为明确患者致病变异并行疾病诊断,对这些家系患者进行全外显子组测序分析,报告如下。
1 对象与方法
1.1 对象
实验研究。2019年12月至2020年12月郑州大学第一附属医院就诊的主诉视力差或失明的先证者(遗传病家系调查中第1 个确诊的患者)且有生育健康后代要求,并检出COL2A1基因致病变异的3个家系(见图1)纳入研究。患者表型采集自门诊病历或本院住院病历。
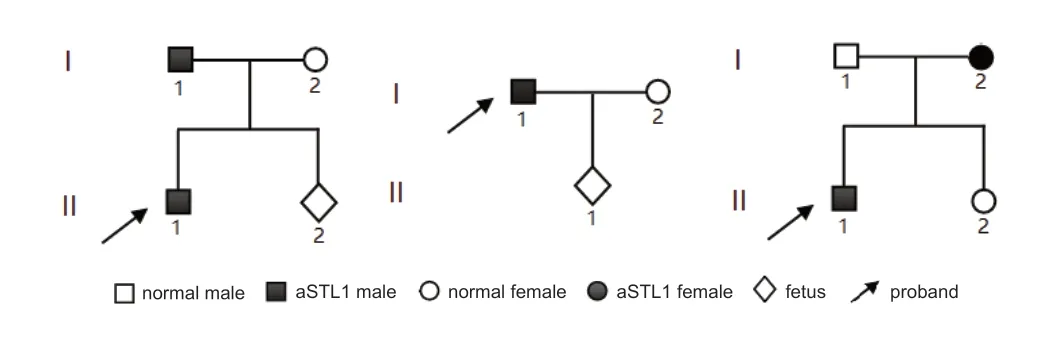
图1.3个非典型Stickler综合征I型家系图Figure 1.Family trees of 3 families with atypical Stickler syndrome I(aSTL1).
家系1先证者(II1),男,5岁。1岁半时发现喜视近物,无眼球震颤,3岁时外院就诊,诊断为“双眼高度近视”,家长发觉其夜间视力稍差,怀疑色盲。否认早产、难产史,否认听力异常,否认关节软骨畸形。双眼检影检查示:右眼-22.00-2.50×35;左眼-21.50-1.75×140。视力检查不合作。眼球前凸,眼前段无明显异常,晶状体透明,眼底呈豹纹状。视网膜电图(ERG)检查显示:双眼视锥、视杆反应重度下降。面部特征:塌鼻梁,小下巴,无腭裂。其父(I1),33岁,高度近视,双眼视网膜脱离(失明),感音神经性听力下降(高频),无腭裂,无骨关节异常,无颅颌面部异常。其母(I2),32岁,健康。
家系2 先证者(I1),男,32 岁,先天性高度近视,双眼视网膜脱离术后,右眼视力丧失,左眼仅剩光感;无腭裂,无全身其他表现。其妻(I2),31岁,健康。
家系3先证者(II1),男,40岁,8岁发病,12岁起视力损害严重,虹膜炎,有双眼视网膜脱离史;大概20岁时已双目失明,白内障,之后无眼科诊疗经历;自述无听力问题,无腭裂,无颅颌面异常,无骨关节异常(见图2)。先证者母亲(I2),66 岁,与先证者(II1)具有相同疾病,现亦失明,无眼科检查及治疗史,并拒绝眼科检查。其父(I1),67岁,健康。其妹(II2),36岁,健康。

图2.非典型Stickler综合征I型家系3患者(II1)颅颌面部外观及眼外观Figure 2.Craniomaxillofacial appearance and ocular appearance of patient (II1) in family 3 with atypical Stickler syndrome I.
家系1孕妇(I2)和家系2孕妇(I2)了解风险并签署知情同意书后,孕18 周时经超声引导下行羊水穿刺取得羊水细胞,不经细胞培养直接用于产前诊断。
本研究严格遵守赫尔辛基宣言,所有受检者签署书面知情同意,获郑州大学第一附属医院伦理委员会批准(批号:KS-2018-KY-36)。
1.2 二代测序
利用Nextera Flex靶向序列富集系统对全外显子组进行磁珠捕获、目标序列扩增,通过Illumina Nova 6000 平台(美国Illumina公司)进行二代测序。捕获序列的平均测序深度为100×,目标序列的覆盖度为97%。人类基因组参考序列版本选择GRCh37。对靶向测序结果注释筛选分析得到候选致病变异位点。可疑致病变异位点的筛选要点为以下几点:①患者疾病表型,如高度近视、失明、白内障、颅面畸形、听力损伤等;②遗传病家族史;③STL1 的临床表现;④了解疾病和致病基因变异的遗传方式,结合患者表型、遗传病家族史,筛选测序所得变异注释信息,进而锁定目标候选变异,⑤对候选变异行致病性分析。最后确认候选致病变异位点。
1.3 Sanger测序
采集家系患者和健康对照者外周血2 ml,乙二胺四乙酸抗凝。使用磁珠法提取试剂盒Blood DNA Midi Kit D3494(美国 Omega bio-tek公司)和自动化DNA提取工作站(Eppendorf epMotion 5075,德国艾本德公司)抽提外周血基因组DNA。以受检者的基因组DNA为模板,使用Taq DNA聚合酶(立陶宛Fermentas公司)和特异引物(GeneTool设计)对COL2A1基因目标位点序列进行扩增。使用纯化柱法(QIAamp DNA Blood Mini Kit,德国QIAGEN公司)提取羊水细胞基因组DNA用于孕妇产前诊断,对相应家系STL1致病变异位点行Sanger测序检测。
PCR条件:95 ℃预变性3 min;95 ℃变性30 s,57 ℃退火30 s,72 ℃延伸45 s,循环30 次;72 ℃终延伸10 min,4 ℃保存。将聚合酶链式反应扩增的目的DNA片段纯化,用荧光标记终止物试剂盒(dGTP BigDye®Terminator,美国ABI公司)和Sanger测序试剂盒(Sequencing Kit,美国ABI公司)进行测序。Sanger测序反应参数:96 ℃预变性1 min;96 ℃10 s,50 ℃ 5 s,60 ℃ 4 min,循环25次,4 ℃保存。采用乙醇/醋酸钠纯化,按BigDyeV3.1操作手册进行。将纯化产物变性后,上样测序仪(ABI 3130 xl测序仪,美国ABI公司)进行序列分析。
1.4 变异致病性分析
检索候选变异在人类基因变异数据库HGMD(Human gene mutation database)、文献数据库PubMed中的收录或报道情况,被专业文献报道过的相关变异为致病性证据支持的变异。通过查询基因组整合数据库GnomAD(The Genome Aggregation Database)了解该变异在人群中出现的频率。参考2015 年版美国医学遗传学和基因组学学院与分子病理学协会(The American College of Medical Genetics and Genomics,ACMG)的描述序列变异的联合共识建议(简称ACMG指南)[4]和《遗传变异分类标准与指南》[5]对候选变异致病性强度等级进行判断。采用GERP++软件和MutationTaster工具对错义变异的氨基酸进行序列保守性分析;利用SIFT、PolyPhen_2、MutationTaster等蛋白预测软件分析错义变异对蛋白质功能的影响,以判断其致病性。
2 结果
2.1 二代测序筛查结果
使用全外显子组测序法对家系1患者(II1)、家系2患者(I1)和家系3患者(II1)进行测序分析,数据分析发现3例患者均检测到COL2A1基因相关致病性变异,见表1。
2.2 候选变异Sanger测序验证
对于全外显子组测序筛查得到的候选基因变异,家系1、2、3的所有患者及家系中的健康成员进行了Sanger测序实验,以验证各个受检者的相关变异携带情况。其中家系1 患者(I1、II1)携带COL2A1基因c.1693C>T(p.R565C)杂合变异,正常健康成员(I2)在该位点无变异,Sanger测序结果见图3。家系2患者(I1)携带COL2A1基因c.2862C>T(p.G954=)杂合变异,正常健康成员(I2)在该位点无变异,Sanger测序结果见图4。家系3 患者(I2、II1)携带COL2A1基因c.2355+1G>A杂合变异,正常健康成员(I1、II2)在该位点无变异,Sanger测序结果见图5。
2.3 变异致病性分析
本研究中无血缘关系的3 个非典型STL1 家系检出3 个候选致病基因COL2A1变异(见表1),这3个COL2A1变异均有国外相关病例携带报道。家系1中患者(I1、II1)COL2A1基因c.1693C>T杂合变异导致II型胶原α 1链第565位氨基酸由极性带正电脂肪族精氨酸(Arg)替换为极性不带电脂肪族半胱氨酸(Cys)(p.R565C),为错义突变,该变异人群中频率为8×10-6,不属于多态性位点;变异位点的氨基酸序列在物种间高度保守,SIFT、PolyPhen_2、MutationTaster均预测COL2A1基因p.R565C变异有害;依据ACMG指南判断COL2A1基因p.R565C变异为致病性(PS1+PS4+PP1+PP3+PP4)。家系2中患者(I1)COL2A1基因c.2862C>T杂合变异为同义突变(p.G954=),无氨基酸序列改变;有研究显示该变异位点会影响COL2A1基因mRNA的可变剪切,导致第42 外显子该变异位点后35 bp序列的删除,进而移码突变产生功能紊乱的蛋白产物[7],该变异在人群中频率极低,GnomAD数据库无记录;依据ACMG指南判断其为致病性(PVS1+PM2+PP4)。家系3中患者(I2、II1)COL2A1基因c.2355+1G>A杂合变异为经典的可变剪接突变,该变异在GnomAD数据库无人群频率记录,依据ACMG指南判断其为致病性(PVS1+PM2+PP4)。
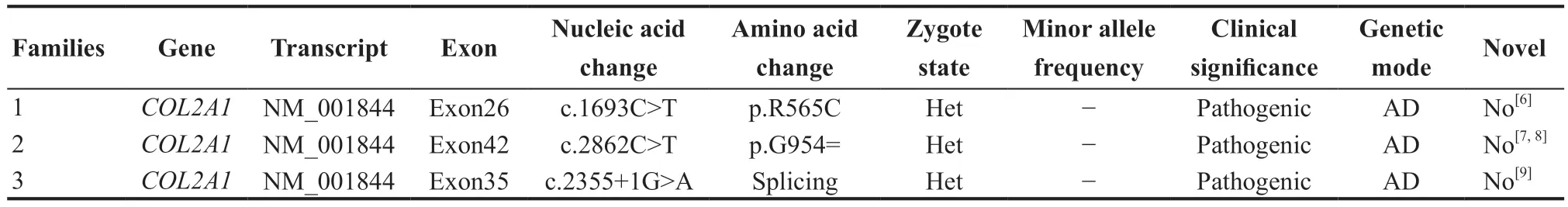
表1.非典型Stickler综合征I型3个家系COL2A1基因变异情况Table 1.Variation of COL2A1 gene in three families with atypical Stickler syndrome type I

图3.非典型Stickler综合征I型家系1 患者(I1、II1)及正常者(I2)COL2A1基因c.1693C>T杂合变异和野生型的Sanger测序图上图红色箭头指向变异所在位置,下图为对应无变异的位置Figure 3.Sanger sequencing results of heterozygous variation and wild type of COL2A1 c.1693C>T in family 1 patients (I1,II1) and normal control (I2) with atypical Stickler syndrome I.The red arrow in the upper figure points to the mutation,the red arrow in the lower figure points to the corresponding site.
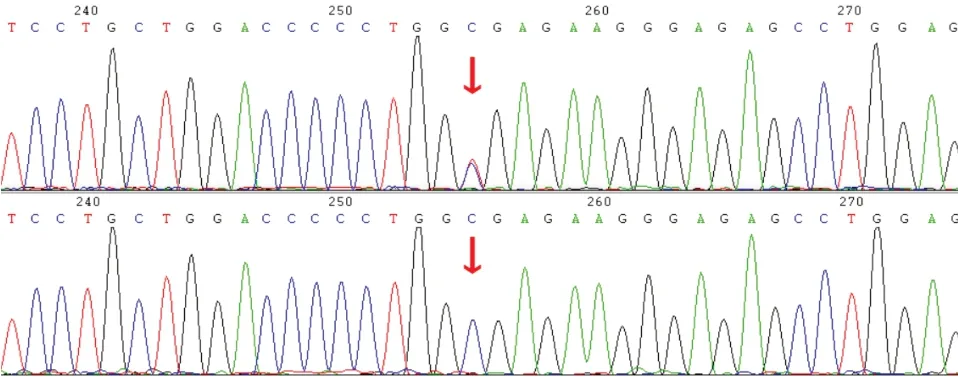
图4.非典型Stickler综合征I型家系2患者(I1)及正常者(I2)COL2A1基因c.2862C>T杂合变异和野生型的Sanger测序图红色箭头指向变异所在位置Figure 4.Sanger sequencing results of heterozygous variation and wild type of COL2A1 c.2862C>T in family 2 patients (I1) and normal control (I2) with atypical Stickler syndrome I.The red arrow in the upper figure points to the mutation,the red arrow in the lower figure points to the corresponding site.
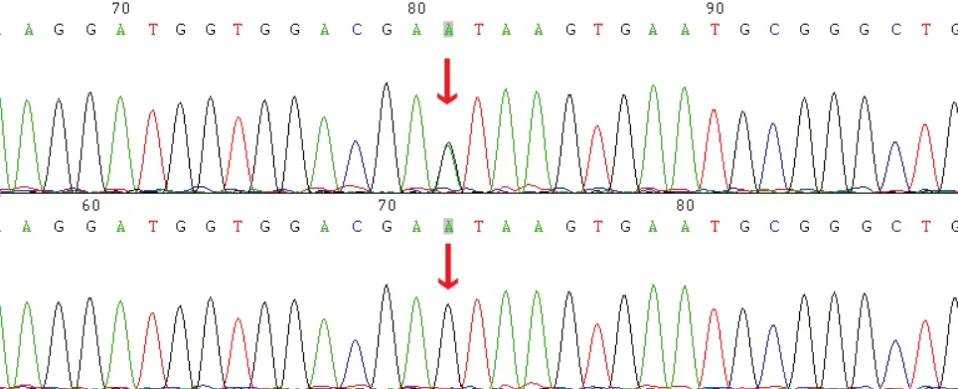
图5.非典型Stickler综合征I型家系3患者(I2、II1)及正常者(I1、II2)COL2A1基因c.2355+1G>A杂合变异和野生型的Sanger测序图红色箭头指向变异所在位置Figure 5.Sanger sequencing results of heterozygous variation and wild type of COL2A1 c.2355+1G>A in family 3 patients (I2,II1) and normal control (I1,II2) with atypical Stickler syndrome I.The red arrow in the upper figure points to the mutation,the red arrow in the lower figure points to the corresponding site.
2.4 Stickler综合征的产前诊断
家系1 孕妇(I2)和家系2 孕妇(I2)分别在孕18 周抽取胎儿羊水进行了COL2A1基因相关位点Sanger测序检测。家系1 中胎儿(II2)未检测到COL2A1基因c.1693C>T(p.R565C)变异,因此该胎儿发展为与先证者(II1)相同基因变异导致的Stickler综合征的可能性小。家系2中胎儿(II1)未检测到COL2A1基因COL2A1基因c.2862C>T变异,因此该胎儿发展为与先证者(I1)相同基因变异导致的Stickler综合征的可能性小。家系1和家系2孕妇都选择了继续妊娠,电话随访出生后情况如下:家系1男孩(II2),2月龄,未见颅颌面异常,新生儿听力筛查通过,眼部及视力未发现异常;家系2 女孩(II1),7月龄,未见颅颌面异常,新生儿听力筛查通过,眼部及视力未发现异常。
3 讨论
本研究收集了3 个非典型STL1 家系患者,他们的病变主要集中在眼部,包括高度近视、视力差或失明、视网膜脱离、白内障,不伴其他异常或伴有其他轻微(轻度)表型。依据孟德尔遗传定律,STL1 为常染色体显性遗传病。针对这3 个家系进行了全外显子组测序分析,通过临床表型结合遗传分析,我们在分子遗传水平帮助这3个家系患者确诊COL2A1基因变异导致的非典型STL1,找到了他们各自的遗传学病因;并对其中2个家系进行了产前诊断,使其获得健康后代。
COL2A1基因编码II型胶原蛋白α1 链,II型胶原蛋白是一种在软骨和眼球玻璃体中发现的纤维胶原。3条完全相同的α1链折叠形成一种左手螺旋的三螺旋分子结构为II型胶原蛋白。COL2A1基因位于染色体12q13.11,由54 个外显子组成,编码1487个氨基酸。三螺旋结构的核心区域(第6─48外显子编码)由一段(甘氨酸-X-Y)n重复序列组成,它对大分子三螺旋结构有重要意义[10]。有研究认为携带位于甘氨酸突变的患者在STL1 中仅约占6%,但常表现出较严重的骨骼系统异常,X和Y位的氨基酸突变的患者骨骼异常较轻[1]。早前有研究认为COL2A1基因2号以外的外显子变异常引起包括眼在内的全身性结缔组织病变及全身性临床特征[11]。COL2A1基因2号外显子发生变异时,STL1主要表现以眼部病变特征为主、无或仅有轻微其他器官组织症状[11,12]。McAlinden等[2]利用小基因构建的体外研究表明,2号外显子变异导致剪接模式的改变和IIA、IIB型mRNA表达率的降低。由于IIA亚型在成人眼玻璃体中表达,眼部表型可能是由于成熟眼中IIA亚型数量不足所致,而改变剪接产生足够的IIB亚型可能使患者缺乏眼外表型。而本研究的3个非典型STL1家系COL2A1基因变异均不在2号外显子区域,这也反映出该病的遗传异质性和该病的复杂性。STL1在国内文献报道较少[13],其中只有部分病例通过分子遗传检测COL2A1基因变异而确诊,如COL2A1基因c.1693C>T(p.R565C)杂合变异的非典型STL1 患者[14]、COL2A1基因c.763-1delG杂合变异的STL1患者[15]。
本研究中3 个非典型STL1 家系患者分别携带COL2A1基因c.1693C>T(p.R565C)、COL2A1基因c.2862C>T(p.G954=)、COL2A1基因c.2355+1G>A致病变异。家系1 中COL2A1基因c.1693C>T(p.R565C)杂合变异导致的患者父子(I1、II1)和国内另1例因该变异导致的患者[14]没有腭裂及严重骨骼问题,他们均为非典型STL1;而国外报道的该变异患者表型严重,包括眼部异常、腭裂、关节问题等,为STL1[6]。p.R565C位于COL2A1基因甘氨酸-X-Y重复序列区的X位氨基酸,因此携带该变异的家系1中的2例患者和国内报道的那例患者表型较轻;携带该变异的国外病例疾病表型较重,这提示相同基因变异的患者临床表现也具有异质性,且基因型和表型之间关系复杂。家系2中COL2A1基因c.2862C>T(p.G954=)杂合变异导致的患者(I1)为非典型STL1,该表型国内未见报道;国外报道的该变异导致的患者为STL1。家系3中COL2A1基因c.2355+1G>A杂合变异导致的患者(II1、I2)为非典型STL1,该表型在国内未见报道;国外该变异导致的患者临床表型有高度近视、中短面部、小下颌、腭裂、皮-罗序列征,听力正常、骨关节正常等[9]。STL1表型复杂,具有遗传和临床表型异质性,本研究显示致病性已知的相同变异导致的Stickler综合征的不同患者表型严重程度也有差异性,这对于STL1的临床特征的认识和诊断非常有意义。临床上非典型Stickler综合征病例较为少见,分子遗传检测可以确诊并明确分型以帮助治疗。缺少基因检测将使该病与其他多种相关疾病的鉴别诊断变得复杂。
本研究从STL1的临床表现和遗传变异水平阐明了3个不同家系的患者的致病原因,分析了相关基因变异的致病性,报道了患者的临床表型。本研究的结果证明了常色体显性遗传非典型STL1遗传异质性及疾病表型的复杂性,为理解该病的分子病理机制提供了更多信息,为非典型STL1的检查确诊、遗传咨询和产前诊断提供了理论依据。由于一些疾病较为罕见,又受到国内不同医院眼科检查设备水平及医师经验所限,分子遗传学检测对非典型STL1 的诊断具有重要的参考价值。同时,利用相应的分子遗传检测技术进行的产前诊断也有效地阻断了非典型STL1向子代传递,帮助患者家庭获得了健康后代。
利益冲突申明本研究无任何利益冲突
作者贡献声明白周现:收集数据,参与选题、设计及资料的分析和解释;撰写论文;对编辑部的修改意见进行修改。邵敬芝:参与设计和修改论文的结果、结论,对编辑部的修改意见进行核修。吴庆华:参与资料的分析和解释,修改论文中结果、结论。孔祥东:参与选题、设计、资料的分析和解释,修改论文中关键性结果、结论
猜你喜欢
中国临床医学影像杂志(2022年2期)2022-05-25
天津医科大学学报(2021年3期)2021-07-21
种子(2021年3期)2021-04-12
外语教学理论与实践(2016年1期)2016-06-11
中国房地产业(2016年9期)2016-03-01
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
郑州大学学报(医学版)(2015年2期)2015-02-27
中华皮肤科杂志(2014年4期)2014-12-19
