
基于加权基因共表达网络分析构建胃癌转移预测模型
2021-09-22 12:07章德昆谢径峰吴芳华黄玉钿薛玉钦王力群
中国癌症杂志 2021年8期
龚 超,陈 魁,章德昆,谢径峰,吴芳华,黄玉钿,薛玉钦,王力群
1.福建医科大学附属福州市第一医院普通外科,福建 福州 350009;2.福建医科大学附属福州市第一医院检验科,福建 福州 350009;3.福建医科大学附属福州市第一医院病理科,福建 福州 350009
胃癌是世界上癌症相关性死亡的主要原因之一,中国2015年胃癌发病率和死亡率中均居癌症发病率和相关死亡率的第二位[1]。尽管目前手术结合化疗的方案已广泛应用于胃癌的治疗当中,但胃癌的5年生存率依然较低[2-3]。据文献[4-5]报道,非转移性胃癌患者5年生存率可达60%,但晚期转移性胃癌患者5年生存率不足10%。因此,早期识别具有高危转移因素的胃癌患者,采取更积极的治疗方案具有重要的临床价值。
近年来基因芯片筛选技术广泛应用于临床治疗,同时有研究[6-7]表明,利用基因芯片技术筛选胃癌转移相关基因具有重要价值。同时每例患者的生物调控机制涉及复杂的基因网络互作,仅通过传统的基因芯片筛选差异基因不可否认其可对生物系统的局部作出解释,但难免遗漏调控过程中的核心分子。本研究利用高通量功能基因组数据库(Gene Expression Omnibus,GEO)中含有晚期转移胃癌数据的数据集(GES103236),采用加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)方法筛选出与胃癌转移相关的靶基因并构建预测模型,拟为胃癌患者个体化诊疗提供参考依据。
1 资料和方法
1.1 GEO数据集获取
本研究从GEO(https://www.ncbi.nlm.nih.gov/geo/)中,获取了微阵列数据集(GSE103236和GSE14210),含有10例转移和非转移的胃癌组织,以及9例癌旁组织,我们使用该数据集作为建模数据集,用于构建基因共表达网络模型。GSE14210含有的123例有疾病进展和预后情况患者用于建立预测模型验证患者疾病进展情况。我们利用Kaplan-Meier plot网站(http://kmplot.com/analysis/)分析多个GEO数据集中胃癌患者的生存数据。同时,含有多种癌症组织和癌旁组织的ONCOMINE数据库(https://www.oncomine.org)也被用于进行外部验证。
1.2 WGCNA
WGCNA是一种常用的模块化分析技术,已被用于识别和筛选复杂疾病的生物标志物或药物靶点[8]。首先,我们对样本的基因表达情况进行质检以及离群值分析,以确保均为样本且能够正常使用。进一步,我们通过R软件中的“WGCNA”分析包构建基因共表达网络[9-10]。然后,通过主要连接关系和皮尔森相关矩阵建立两个基因之间的相关矩阵。并通过对网络拓扑结构的分析,确定软阈值大小。软阈值是基于近似无标度拓扑的一种准则。无标度拓扑拟合指数曲线在达到较高值后趋于平缓,选择的软阈值还需要有一个更好的平均连通性。我们进一步将邻接转化为拓扑重叠矩阵(topological overlap matrix,TOM),TOM可以度量一个基因的网络连通性,该基因定义为其与其他所有基因邻接的总和,用于网络生成[11-12]。为了将表达谱相似的基因分类到基因模块中,根据基于TOM的差异测度进行平均连锁层次聚类[9-10]。模块鉴定后,根据各组表型数据,采用t检验计算各组间各基因表达显著性检验的P值。每个模块的显著性定义为模块内基因显著性的平均值。具有显著性的模块可能与特定疾病的存在有关。为了进一步分析模块,我们计算模块特征基因(MEs)的差异性,为模块树状图选择一条切线,并合并部分模块。
1.3 特征模块和核心基因筛选
在对每个基因模块进行主分析时,MEs被视为主要成分,所有基因的表达模式可归纳为给定模块内的单一特征表达谱。另外,我们通过皮尔森相关检验来评估MEs与转移的相关性,以确定相关模块。选择与转移高度相关的模块作为转移模块进一步进行分析。此外,为了确定与转移相关的模块,我们进行了模块与转移之间的皮尔森相关性分析。识别中心的基因,我们选择转移模型相关系数最高的数据集,该模块也是在所有的模块中比重最大,在模块和中心基因定义的模块连接以绝对值衡量皮尔森的相关性。为了寻找真正的核心靶基因,我们对数据集进行了差异基因分析,同时利用VEEN图对癌组织和癌旁组织的差异基因、转移和非转移的差异基因和Lightsteelblue模块中基因取交集。我们筛选出了C5AR1、AP3M2、TYMP、ANXA2P1用于进一步分析。
1.4 基因本体(gene ontology,GO)功能学和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路分析
为了解Lightsteelblue模块主要涉及的功能学和通路。我们采用标准富集计算方法进行GO功能分析和KEGG通路分析用以筛选与其相关的功能和通路。
1.5 统计学处理
统计分析采用的是SPSS 24.0,GraphPad Prism 7.0和R 3.4.1,同时用上述软件进行图像生成处理。t检验用来分析两个组别之间的平均数的差异。绘制受试者工作特征(receiver operating characteristic,ROC)曲线,评估核心靶基因的预测能力,利用曲线下面积(area under curve,AUC)评估灵敏度和特异度。利用Kaplan-Meier法绘制生存曲线,预测基因对病患预后的影响。P<0.05为差异有统计学意义。
2 结果
2.1 差异基因分析
利用“edge”R筛选差异表达的基因,基于fold change=1(P<0.05)为阈值筛选GSE103236数据集中癌组织和癌旁组织差异基因,共发现3 431个差异表达基因,其中2 399个基因表达上调,1 032个基因表达下调。同理,对转移和非转移患者进行差异基因筛选,共发现1 264个差异表达基因,其中1 218个基因表达上调,46个基因表达下调。同时,通过R软件绘制热图,50个基因在10个癌组织、9个癌旁组织、7个非转移癌组织和3个转移癌组织中的表达情况见图1。

图1 热图分析 Fig.1 Thermographic analysis
2.2 构建共表达模块
基于19 711个差异基因在10例胃癌组织和9例癌旁组织的表达数据,利用基于无序列网络的WGCNA方法将基因进行模块化富集分析(图2),将基因依据其各相关表达量进一步进行分类。为此,共筛选获得了79个相应的基因模块,为了进一步了解各模块与转移的关联度,本研究进一步将临床信息纳入分析,检测各模块与转移的皮尔森相关系数,以便筛选出与转移最相关的模块,纳入进一步的分析。我们依据各模块在转移上皮尔森系数绝对值相加为最高者认定为响应系数最高模块,最后筛选获取了Lightsteelblue模块。

图2 WGCNA模式图Fig.2 WGCNA cluster dendrogram
2.3 基因模块与临床性状关联分析
为寻找与转移相关的关键调控基因及效应通路,基于GO功能富集以及KEGG数据库中信号转导通路的上下游关系,本课题组依据Lightsteelblue模块中所含有的47个相关基因筛选表达响应基因参与的信号通路的关联图(图3A、B)。我们据此发现多条极为相关的信号转导通路,如细胞黏附、转录异常调控等。目前已有较多的研究[13-15]显示,上述通路在胃癌中涉及转移敏感性。因此初步证明了本课题前期中所筛选出的Lightsteelblue模块是和转移相关的模块。同时,为了进一步验证该模块是否与转移相关,我们将Lightsteelblue模块中基因与转移一起予以分析(图4),结果显示,在Lightsteelblue模块中大部分基因其P值均小于0.05,进一步佐证了Lightsteelblue模块与转移的相关性。基于此我们可以初步认定Lightsteelblue模块就是与转移关联最大的模块。为进一步确保模块中的基因与胃癌发生、发展及转移相关,我们进一步通过VEEN分析,对模块中的基因和我们前期所筛选的差异基因取交集。最终,我们共筛选了4个基因C5AR1、AP3M2、TYMP、ANXA2P1作为核心靶基因(图5)。

图3 GO分析和KEGG分析Fig.3 GO analysis and KEGG analysis
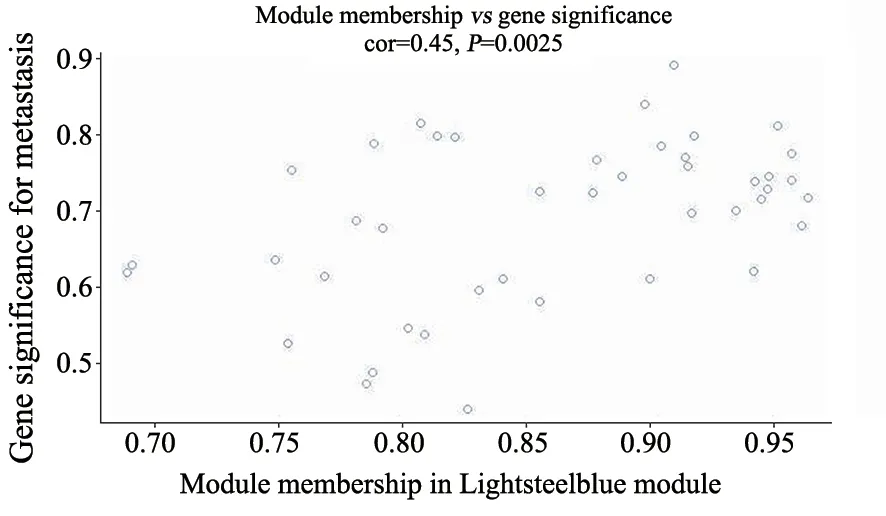
图4 Lightsteelblue模块基因与转移相关分析Fig.4 Analysis of the relationship between Lightsteelblue module gene and metastasis

图5 VEEN图Fig.5 VEEN
2.4 核心靶基因验证
为了进一步验证我们筛选的基因对预测患者胃癌是否转移以及预后的效能,我们重新纳入原始数据集进一步分析。本课题前期筛选的4个基因表达在肿瘤组织中表达显著高于癌旁组织(P<0.01,图6),同时在转移组中,其表达量显著高于非转移组。进一步证明了我们前期筛选的基因不仅与胃癌转移相关,而且为明确的癌基因。为验证基因表达丰度能否判定肿瘤的发生或者转移,采用了ROC曲线计算其AUC用于判定各基因在预测肿瘤发生以及预测转移的概率。
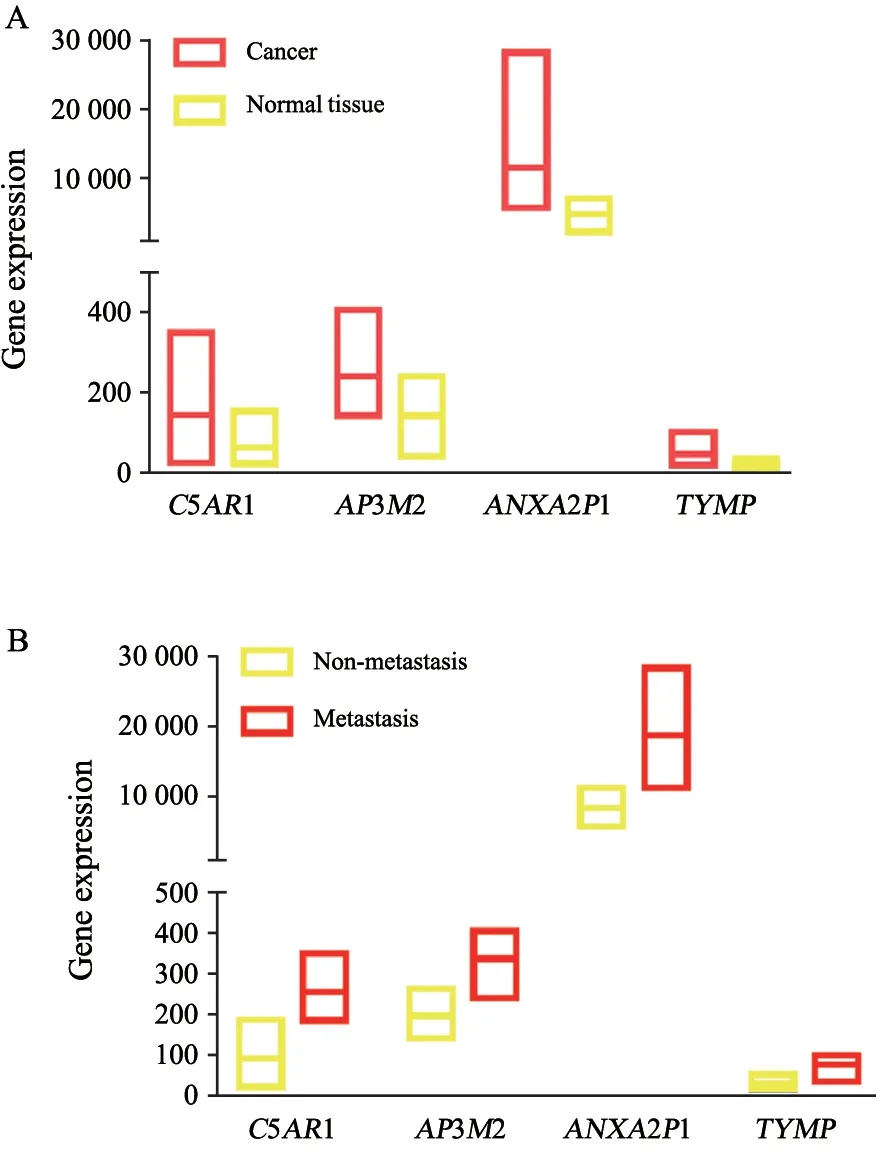
图6 各基因相对表达量Fig.6 The relative expressions of different genes
在预测肿瘤的发生中,C5AR1、AP3M2、TYMP、ANXA2P1基因其分别预测概率为0.767、0.844、0.956和0.867。基于此我们进一步建立了回归预测模型,该模型预测肿瘤发生其AUC可达1。上述结果提示前期所筛选的基因在预测肿瘤发生中也有较大的意义。为进一步验证其用于预测胃癌转移的效能,在预测胃癌转移发生中,C5AR1、AP3M2、TYMP、ANXA2P1基因其分别预测概率为0.952、0.905、0.952和0.857。基于此我们进一步建立了LOGSTICAL预测模型,该模型预测肿瘤发生其AUC可达1。上述结果说明我们所筛选的基因与胃癌发生、发展和转移相关(图7)。
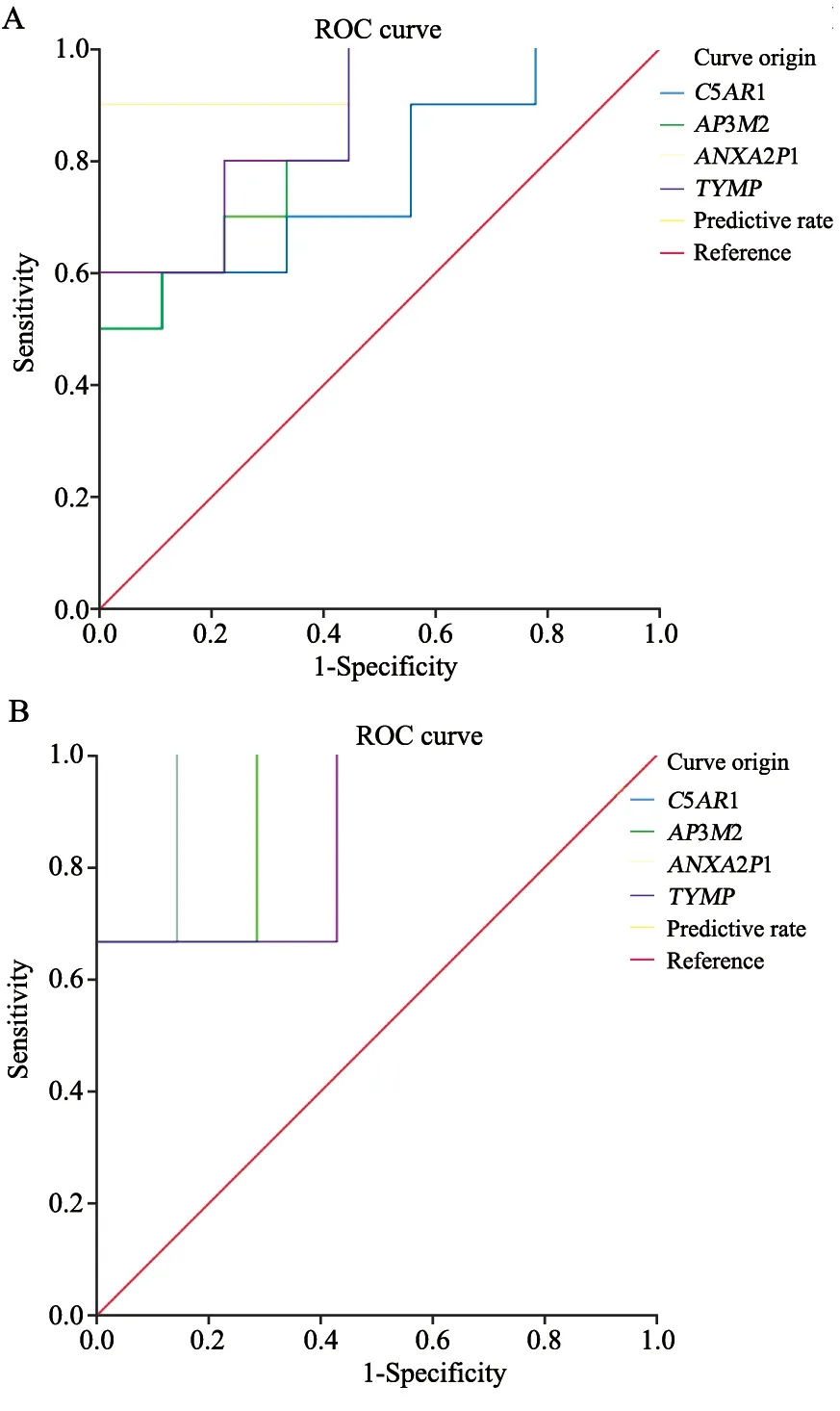
图7 ROC曲线预测肿瘤发生和转移 Fig.7 The prediction of tumor genesis and metastasis by ROC curves
2.5 核心靶基因外部表达和生存验证
我们利用ONCOMINE数据库中包含有大量的mRNA测序/芯片结果,对C5AR1、AP3M2、TYMP和ANXA2P1基因多数据集的meta分析,在所纳入的8个数据集中,所筛选的C5AR1、AP3M2、TYMP和ANXA2P1基因在癌组织中均相对癌旁组织呈现高表达(P=0.002,P=0.001,P<0.001,P=0.003,图8)。结果表明我们的前期结果是可信的。同时我们利用Kaplan-Meier plot网站(http://kmplot.com/analysis/)所含有的多个胃癌生存数据集分析我们所筛选的基因是否可以预测预后。结果显示,在胃癌患者中,C5AR1、AP3M2、TYMP和ANXA2P1基因低表达患者有着更好的预后,进一步佐证了我们前期结果(图9)。

图8 ONCOMINE数据库分析图Fig.8 ONCOMINE database analysis

图9 Kaplan-Meier plot数据库分析生存情况Fig.9 Survival curves by Kaplan-Meier plot database analysis
2.6 利用GSE14210构建核心基因预测复发和生存模型
为进一步明确所筛选的基因是否能有效地预测胃癌患者的复发及生存情况,我们利用生存风险模型分析GEO数据库中带有总生存以及疾病进展资料的数据库GSE14210。其内共有167例胃癌患者基因芯片数据,有123例患者带有确切的疾病进展以及生存资料数据。本研究利用该数据集,通过对上述核心基因表达数据进行提取以及生存数据分析,构建生存风险预测模型(图10),其公式=C5AR1×(0.113 8)+AP3M2×(0.000 1)+TYMP×(0.093 9)+ANXA2P1×(0.486 3)。据此,我们可以获得每个患者的风险得分,并利用该得分对患者是否出现疾病进展进行预测,结果表明,所构建的生存风险模型能较好地预测胃癌患者治疗后是否出现疾病进展(AUC=0.71,P<0.001,图11)。据此有理由相信我们所筛选的基因以及模型是可靠的。

图10 构建生存风险模型Fig.10 Survival risk model
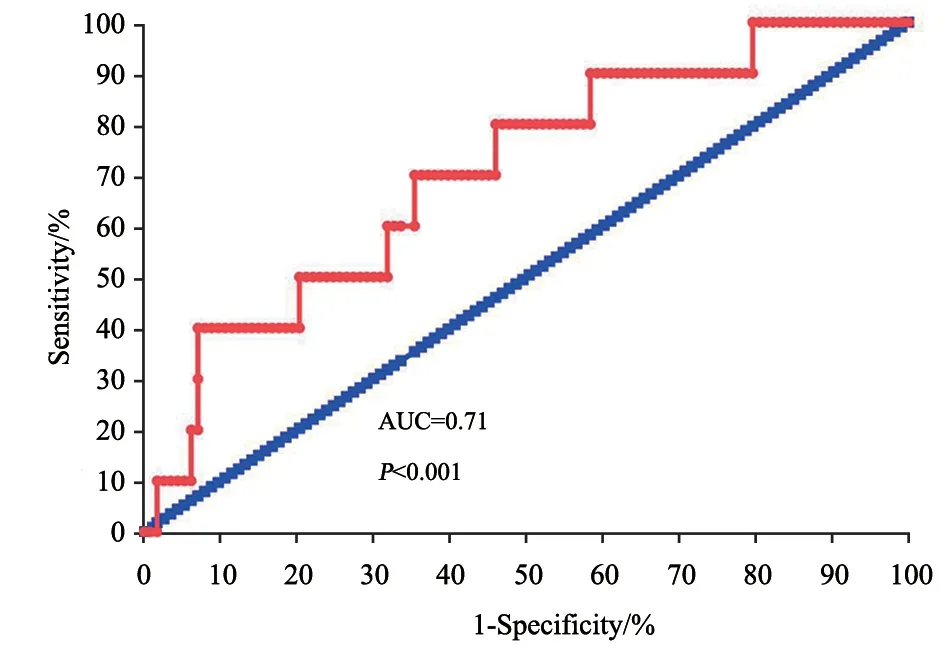
图11 ROC曲线Fig.11 ROC curve
3 讨 论
胃癌是世界上癌症相关性死亡的主要原因之一,尽管目前手术结合化疗/免疫治疗/靶向治疗等多种方案已广泛应用于胃癌的治疗当中,但胃癌的5年生存率依然低下[2-3]。这主要是由于大多数胃癌被发现时已处于中晚期,所以预后不佳。晚期转移性胃癌患者5年生存率不足 10%[4-5],因此,本研究为更早识别具有高危转移风险的胃癌患者,对GEO数据集转移和非转移胃癌组织及癌旁组织的数据集,通过精准的WGCNA算法识别出了4种与胃癌发生、转移相关的基因,即C5AR1、AP3M2、TYMP和ANXA2P1;同时通过内部表达验证和利用外部的ONCOMINE数据库和Kaplan-Meier plot数据库验证了我们所筛选的基因具有较强的重复性和可靠性。
WGCNA通过将具有相似的功能或者出现在同一生物通路中的基因视作一个模块,进而在基因模块水平上研究每个生物个体与外部信息的联系。并通过有效地划分基因模块进一步研究生物调控网络内部关联性[8]。因此基于WGCNA的方法构建基因网络互作关系模式,不仅可以帮助我们从基因网络调控角度更为精准地筛选出核心靶基因,又可避免由于患者个体基因表达差异导致的假阳/阴性结果。本研究利用该方法,筛选出了C5AR1、AP3M2、TYMP和ANXA2P1基因。同时,我们通过表达分析发现,这些基因在胃癌组织中相较于癌旁组织呈高表达,在转移患者中表达量较非转移患者高。上述结果说明我们通过WGCNA方法筛选的基因是有效的。为了在外部数据库中验证,我们利用了ONCOMINE数据库,其包含多个胃癌和癌旁组织测序/基因芯片的结果,通过对多个数据集进行的meta分析发现,上述4个基因在胃癌组织中相较于癌旁组织均高表达。同时,我们还通过包含多个不同来源数据集胃癌患者生存数据的Kaplan-Meier plot网站,验证了C5AR1、AP3M2、TYMP和ANXA2P1基因与患者预后的关系。结果显示,上述4个基因高表达均预示着患者有不良预后。同时,本研究还利用了GSE14210数据集,基于上述的核心靶基因构建了疾病进展和生存模型,该模型的结果也能较好地预测患者疾病进展,从而进一步佐证了我们所筛选的核心基因可以有效地预测患者的疾病进展和预后。因此,本研究结果对未来阐明C5AR1、AP3M2、TYMP和ANXA2P1基因表达与胃癌转移及预后的关系奠定了一定的基础。但不可否认的是,本研究的结果还需要在大量临床实际样本中进一步验证,并通过一系列的体内/体外实验探索上述4个基因的作用、相互关系、机制及临床意义。
总之,C5AR1、AP3M2、TYMP和ANXA2P1基因有可能成为新的预后指标,有助于胃癌患者个性化治疗及临床预后判断。
猜你喜欢
心电与循环(2020年1期)2020-02-27
江苏农业科学(2017年5期)2017-04-15
财经(2017年2期)2017-03-10
财经(2016年15期)2016-06-03
财经(2016年3期)2016-03-07
财经(2016年6期)2016-02-24
中国卫生标准管理(2015年3期)2016-01-14
医学研究杂志(2015年9期)2015-07-01
中国当代医药(2015年20期)2015-03-01
湖北农业科学(2014年3期)2014-07-21
