
结合珠蛋白通过抑制ERK1/2减轻肝细胞铁死亡
2021-09-09 08:09张颖婷林龙帅何恩俊何咏元苏滢泓段澄澄王斯源赵庆华
上海交通大学学报(医学版) 2021年8期
魏 倩,张颖婷,林龙帅,,何恩俊,何咏元,苏滢泓,段澄澄,王斯源,赵庆华,赵 倩,3,贺 明
1.上海交通大学医学院病理生理学系,细胞分化与凋亡教育部重点实验室,上海 200025;2.上海交通大学附属第一人民医院骨科,上海 201620;3.上海交通大学应激与肿瘤创新单元(2019RU043),中国医学科学院,上海 200025
铁死亡(ferroptosis)是一种由大量脂质过氧化产物和活性氧(reactiveoxygen species,ROS)堆积而导致的铁依赖的调控性细胞死亡[1-2]。在生化特征上,铁死亡主要表现为铁沉积和脂质过氧化[3-4],其中铁沉积通过芬顿反应产生高活性的羟基自由基或激活脂氧合酶来促进氧化损伤[4]。同时,氧自由基与生物膜上的磷脂以及多不饱和脂肪酸等分子发生脂质过氧化反应,形成大量的脂质过氧化产物,如4-羟基壬烯醛(4-hydroxynonenal,4-HNE)和丙二醛(malondialdehyde,MDA),从而使细胞膜的流动性和通透性发生改变,进而导致细胞发生损伤[5]。同时,铁死亡诱导剂可以引起基因表达的改变从而诱导细胞脂质过氧化或死亡,包括前列腺素内过氧化物合酶2(prostaglandinendoperoxide synthase2,PTGS2)[3]和酰基辅酶A合成酶长链家族成员4(acyl-CoA synthetaselong-chain family member 4,ACSL4)[6]表达明显增加,谷胱甘肽过氧化物酶4(glutathioneperoxidase4,GPX4)表达下调。
铁死亡在多种肝脏疾病的发生与发展中发挥重要作用[7]。在酒精性肝病(alcoholic liver disease,ALD)的研究中,酒精饮食诱导肝脏铁死亡,而且,酒精性肝损伤可以显著被铁死亡抑制剂Ferrostatin-1(Fer-1)所抑制[8-9]。近年来的研究[10]发现,铁代谢紊乱引起的细胞内铁超载是非酒精性脂肪性肝病(nonalcoholic fatty liver disease,NAFLD)发展为非酒精性脂肪性肝炎(nonalcoholic steatohepatitis,NASH)及NASH加重的病因之一,提示铁死亡在NASH发生发展中发挥了重要的作用。而且,多种药物可以通过调节相应的基因表达诱导肝星状细胞(hepatic stellatecells,HSCs)发生铁死亡从而抑制肝纤维化的发展[11]。而单纯长期的高铁饲料饲喂是常用的铁死亡动物模型[12-14],高铁饲料可以使肝脏发生铁沉积,进而引起肝细胞铁死亡的发生。研究[12]表明,由高铁饮食诱导的小鼠肝细胞死亡可以被铁死亡抑制剂Fer-1和去铁胺(deferoxamine,DFO)显著缓解,而其他细胞死亡形式的抑制剂没有这种效果。目前高铁饮食导致肝细胞铁死亡的机制仍不明确,探寻新的介导肝细胞铁死亡的信号分子和机制对于肝病治疗非常必要。
结合珠蛋白(haptoglobin,Hp)是肝脏合成的一种急性期时相反应蛋白(acute phase protein,APP)。作为主要的血浆蛋白之一,Hp蛋白的主要功能是与游离血红蛋白(hemoglobin,Hb)结合成稳定复合物,使肝脏回收血红素铁进行代谢,以防止肾脏受损[15]。同时Hp多态性可引起血红蛋白结合能力、抗氧化和铁循环活性的改变,在细胞的氧化应激反应中具有重要作用[15]。临床研究[16]发现,严重肝病时血清Hp表达水平降低。而且Hp也可以作为早期预测和诊断肝硬化患者发生肝细胞癌的潜在标志物[17]。但是Hp在正常肝细胞发生铁死亡过程中是否发挥作用以及作用的机制尚未见报道。
本研究结合RNA测序(RNA-sequencing,RNA-seq)技术和生物信息技术,利用高铁饮食小鼠模型和铁死亡诱导肝细胞实验探寻新的参与肝细胞铁死亡的信号分子的表达、作用及机制。
1 材料与方法
1.1 实验动物及细胞
C57BL/6J品系小鼠购买于上海灵畅生物科技有限公司,在上海交通大学医学院的实验动物科学部SPF(specific pathogen-free)级动物房中饲养。使用许可证号为SYXK(沪)2018-0027,生产许可证号为SCXK(沪)2018-0007。小鼠进食和饮水自由,每笼5只小鼠。动物房温度18~26℃,相对湿度40%~60%,昼夜交替时间各12 h。所有实验动物相关操作均已经过上海交通大学医学院实验动物使用和管理委员会批准。
正常小鼠AML-12肝细胞购自美国模式菌种保藏中心(American Type Culture Collection,ATCC)。
1.2 主要试剂和仪器
AIN76A高铁饲料(Dyets公司,美国),RAS合成致死分子3(RAS-selective lethal small molecule 3,RSL3)、SCH772984(Selleck公司,美国),C11 BODIPY581/591(Thermo Fisher公司,美国),horchest、MDA检测试剂盒(上海碧云天生物技术有限公司),Hp抗体(Abcam公司,英国),细胞外信号调节激酶1/2(extracellular regulated protein kinases 1/2,ERK1/2)抗体、磷酸化的ERK1/2(phosphorylated ERK1/2,p-ERK1/2)抗体(CST公司,美国),相应辣根过氧化物酶(horseradish peroxidase,HRP)标记的β-微管蛋白(β-tubulin)抗体(GNI公司,日本),HRP标记的二抗anti-rabbit IgG(Santa Cruz公司,美国),4-HNE抗体(ADI公司,美国),铁(ferrum,Fe)含量检测试剂盒、谷丙转氨酶(glutamic-pyruvic transaminase,GPT)检测试剂盒、谷草转氨酶(glutamic-oxalacetic transaminase,GOT)检测试剂盒(南京建成生物工程研究所有限公司),TRIzol、lipofectamine 2000转染试剂(Invitrogen公司,美国),AMV反转录酶(TaKaRa公司,日本),10%胎牛血清、DMEM/F12培养基(Gibco BRL公司,美国),ITS液体培养基补充剂(100×)(Sigma公司,美国)。
LX-20全自动生化分析仪(Bio-Tek公司,美国),LAS 4000 mini化学发光成像仪(富士公司,日本),BD FACSAria流式细胞仪(BD Biosciences公司,美国),激光共聚焦显微镜(Nikon公司,日本)。
1.3 高铁饲料饮食诱导肝细胞铁死亡小鼠模型的构建
体质量20~25 g的C57BL/6J品系雄性小鼠,8周龄,共13只。随机分为2组,分别是正常饲料喂养的小鼠(normal iron diet,NID组,n=5)和高铁饲料(8.3 g羰基铁/kg)喂养的小鼠(high iron diet,HID组,n=8)。连续喂食12周,实时监测小鼠饮食饮水量和体质量变化。12周后处死小鼠取血和肝脏。
1.4 小鼠肝脏病理学观察
将4%多聚甲醛固定的肝组织包埋于石蜡中。石蜡切片后分别进行苏木精-伊红(hematoxylin-eosin,H-E)染色、普鲁士蓝染色和天狼星红染色,脱水封片,在光学显微镜下观察并拍照。
1.5 肝损伤血清学生化指标检测
将收集至肝素抗凝管的小鼠全血4℃下587×g离心20 min,取上层血清至干净EP管中并做好标记。利用GPT和GOT检测试剂盒(96T)检测小鼠血清中的GPT和GOT水平,测量510 nm处的反应孔吸光度值,计算血清样品中GPT和GOT的水平。
1.6 免疫组织化学法检测肝组织中4-HNE的表达
将包埋在石蜡中的肝组织切片进行脱蜡水化、修复抗原和封闭后,用4-HNE抗体4℃孵育过夜,随后加入二抗,室温孵育30 min。二氨基联苯胺(diaminobenzidine,DAB)染色,苏木精复染,乙醇梯度脱水,二甲苯透明,环氧树脂封片,在光学显微镜下观察4-HNE表达,肝细胞胞质显示棕黄色即为阳性细胞。
1.7 肝组织中MDA含量检测
切取小块肝组织(30 mg)称重并记录,置于2 mL EP管内,根据肝组织质量加入1×PBS(每10 mg肝脏组织加入100μL PBS)。加入干净的磁珠2颗,置入提前预冷的匀浆器内匀浆(强度为60 Hz,时长为2.5 min,模式为运行10 s、停止10 s)。最大转速离心后将匀浆后的液体吸入干净的1.5 mL EP管内,4℃、12 000×g离心10 min,取上清液置于干净1.5 mL EP管内,后续检测步骤根据MDA试剂盒说明书进行。
1.8 RNA抽提、反转录及实时荧光定量PCR实验
肝组织和AML-12(alpha mouse liver 12)肝细胞用TRIzol进行裂解,按照标准操作方法抽提RNA,定量后利用AMV反转录酶将mRNA反转录成cDNA用于实时荧光定量(real-time)PCR实验。采用Power SYBPgreen PCR Master Mix荧光定量试剂盒,针对待测的目的基因设计PCR引物,引物由上海生工生物工程有限公司合成,引物序列见表1。每个待测样品设置3个复孔以防实验误差。内参基因选择小鼠核糖体蛋白(ribosomal protein L13a,Rpl13a),采用2-ΔΔCT计算每个基因的相对表达量。

表1 Real-time PCR引物序列Tab 1 Primer sequencesfor realtime-PCR
1.9 肝组织样本RNA测序

1.10 蛋白质印迹法(Western blotting)
抽提小鼠肝脏蛋白和AML-12肝细胞蛋白后,根据待测蛋白的相对分子质量大小利用相应的8%~15%SDS-聚丙烯酰胺凝胶进行电泳,并转移至硝酸纤维素膜上,随后用5%的脱脂牛奶室温封闭1 h,4℃孵育Hp抗体(1∶500)或p-ERK1/2抗体(1∶500)或ERK1/2抗体(1∶500)或β-tubulin-HRP抗体(1∶5 000)在摇床上振荡过夜。回收一抗,TBST缓冲液洗膜3次,每次5 min。HRP标记的二抗anti-rabbit IgG(1∶5000)室温摇床孵育1 h,TBST缓冲液洗膜3次,每次5min。将含有HRP底物的显影液滴在膜上,LAS4000 mini成像仪拍照并保存。
1.11 Hp过表达和Hp siRNA转染实验
AML-12细胞使用补充有10%胎牛血清、40 ng/mL地塞米松和5 ng/mL ITS的DMEM/F12培养基在37℃、5%CO2恒温箱中进行培养。待细胞密度达到50%~70%后使用lipofectamine 2000转染试剂进行HpsiRNA或Hp质粒转染,按照说明书推荐的用量和步骤进行,6 h后换成新鲜的完全培养基继续培养,所用siRNA序列见表2。48 h后将上述细胞接种至6孔板和12孔板中,给予终浓度为5μmol/L的RSL3处理,刺激24 h后收集细胞样品。
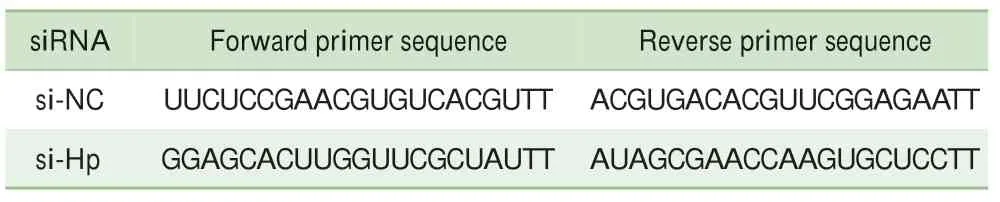
表2 所用siRNA序列Tab 2 Sequencesof siRNA
1.12 ERK1/2介导Hp作用实验
ERK1/2介导Hp作用实验前序步骤与Hp过表达和HpsiRNA转染实验相同,在给予RSL3之前1 h给予终浓度为5μmol/L的SCH772984,刺激24 h后收集细胞样品,进行后续实验。
1.13 肝细胞C11-BODIPY 581/591流式检测
在AML-12肝细胞培养上清液中加入C11-BODIPY581/591,终浓度为5μmol/L,37℃孵育30 min。对标记细胞用胰蛋白酶37℃孵育4 min,再重悬于完全培养基中,流式细胞术检测每个样本细胞的荧光强度。每个条件至少分析10 000个细胞。
1.14 肝细胞C11-BODIPY 581/591免疫荧光检测
在细胞培养基上清液中加入C11-BODIPY581/591,终浓度为5μmol/L,37℃孵育30 min,随后10μg/mL horchest 37℃孵育5 min,用PBS洗涤2次,去除多余的C11-BODIPY581/591,在激光共聚焦显微镜下拍照观察。
1.15 统计学分析
所有数据统计分析采用Microsoft Excel和GraphPad Prism 6.0软件计算处理,连续性定量数据用±s表示,采用Student′st检验进行2组间比较,以P<0.05判定为差异具有统计学意义。
2 结果
2.1 高铁饮食诱导小鼠肝细胞铁死亡和肝损伤
NID组小鼠和HID组小鼠在饲喂12周过程中体质量及摄食量均无明显差异,2组小鼠肝脏质量/体质量无明显差异(图1A、B)。但相较于NID组小鼠,HID组小鼠肝脏的边缘较钝,呈不规则凹陷且质地较硬(图1A)。同时,肝脏H-E染色和天狼星红染色显示,NID组肝脏细胞形态正常,未见明显肝纤维化,而HID组小鼠肝脏组织出现明显的肝细胞坏死及肝纤维化(图1C、D)。而且,血清学生化指标检测结果显示,HID组小鼠血清GPT和GOT水平较NID组显著增高(图1E、F),以上提示高铁饮食可以明显诱导小鼠发生肝损伤和纤维化。为了验证高铁饮食引起的小鼠肝损伤是否与铁死亡有关,我们比较了2组小鼠肝脏的铁沉积和脂质过氧化水平。肝脏普鲁士蓝染色(图1G)和肝脏铁含量检测(图1H)均显示,HID组小鼠肝脏出现了明显的铁沉积。4-HNE免疫组织化学检测发现,NID组小鼠没有4-HNE累积,而HID组小鼠肝脏中4-HNE累积明显(图1I)。同时,HID组小鼠的肝脏中MDA的含量明显高于NID组(图1J)。此外,PTGS2、ACSL4和GPX4作为铁死亡发生过程中的重要参与分子,其mRNA的表达是评价是否发生铁死亡的重要指标。real-time PCR结果显示,HID组小鼠肝脏中PTGS2和ACSL4mRNA表达水平明显上升(图1K、L),而GPX4mRNA的表达水平受到了明显抑制(图1M)。以上结果提示高铁饮食可以诱导小鼠肝脏发生铁沉积和脂质过氧化从而引起铁死亡。
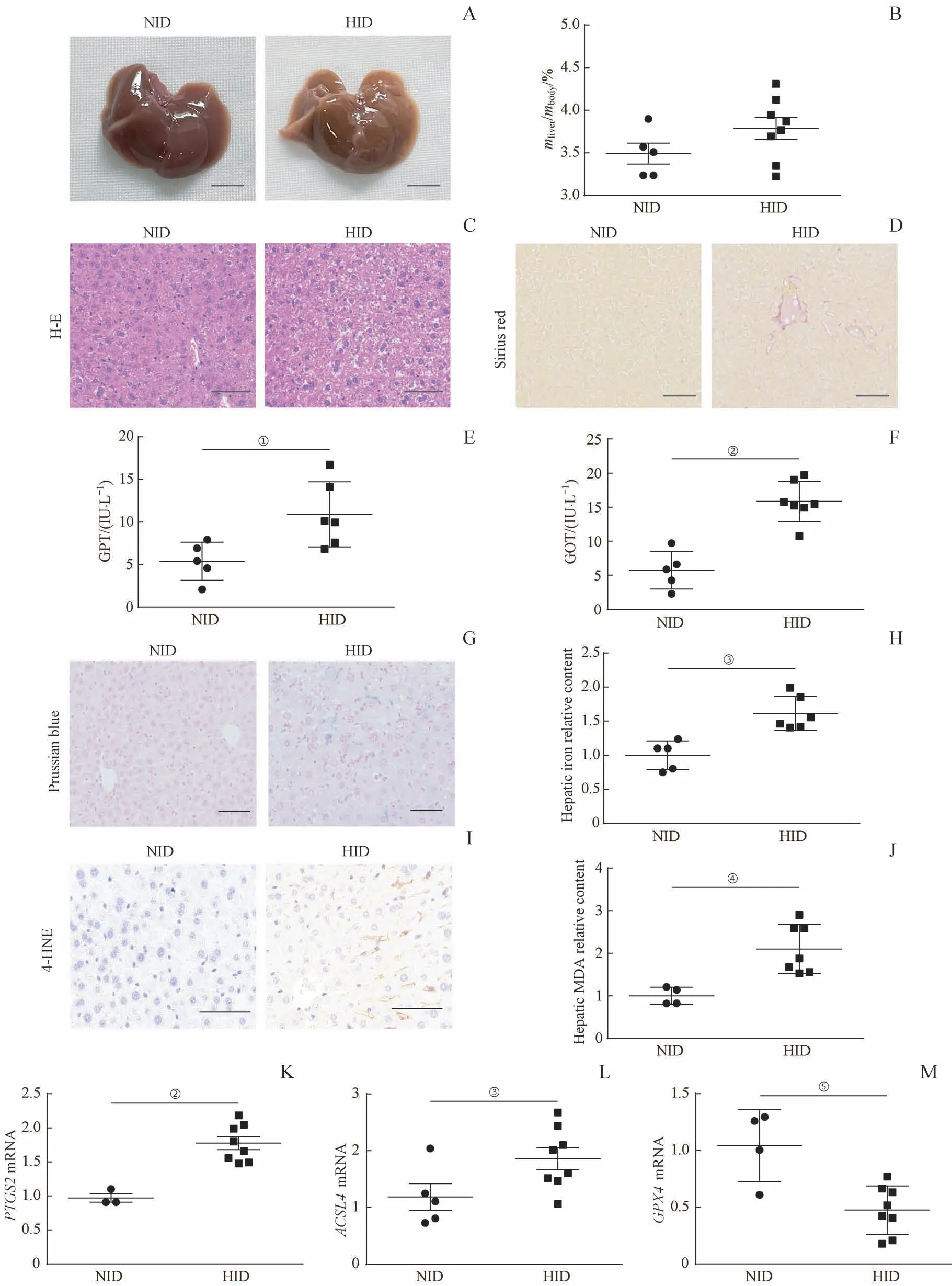
图1 高铁饮食诱导小鼠发生肝细胞铁死亡和肝损伤Fig 1 High iron diet-induced hepatocyte ferroptosis and liver injury in mice
2.2 高铁饲料饮食对小鼠肝脏转录组表达的调控

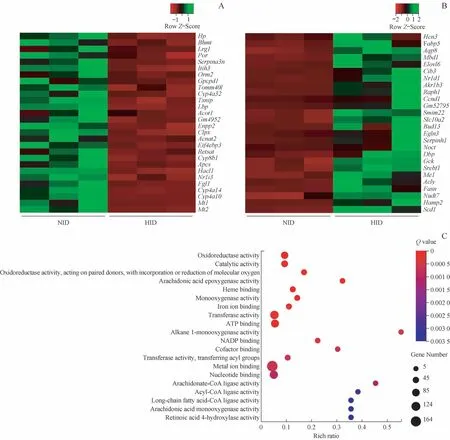
图2 高铁饮食引起的小鼠肝脏中转录组的表达变化Fig 2 Dysregulated mRNAs in livers of micefed with high iron diet
2.3 Hp在肝细胞铁死亡中表达减少
进一步通过GSEA分析发现HID组中急性时相反应被明显抑制,但是其在NID组得到明显富集且富集评分(enrichment score,ES)排名第一(图3A)。其中,表达下调的Hp均参与了GO和GSEA分析中发生调节变化的通路(图3B),因此推测Hp可能参与了高铁饮食导致的肝细胞铁死亡。利用real-time PCR和Western blotting分别检测了小鼠肝组织中Hp的mRNA和蛋白水平,发现相比NID组,HID组小鼠肝脏中Hp的mRNA和蛋白水平均明显下调(图3C、D),这与RNA-seq结果一致。给予AML-12肝细胞铁死亡诱导剂RSL3后Hp的mRNA和蛋白表达水平也同样显著下调(图3E、F)。铁死亡发生过程中Hp的表达受到明显抑制,提示Hp可能在肝细胞铁死亡中发挥作用。

图3 高铁饮食和RSL3抑制肝细胞Hp的mRNA和蛋白表达Fig 3 High iron diet and RSL3 inhibiting theexpression of Hp mRNA and protein in hepatocytes
2.4 Hp过表达抑制RSL3诱导的肝细胞铁死亡
为了验证Hp是否在肝细胞铁死亡中发挥作用,我们在正常小鼠肝细胞AML-12细胞中转染Hp质粒48 h后,给予铁死亡诱导剂RSL3刺激24 h。Real-time PCR和Western blotting结果显示,转染Hp质粒后,AML-12肝细胞内的HpmRNA和蛋白表达水平显著上调(图4A、B)。细胞计数结果显示,给予RSL3刺激后细胞死亡增加,细胞数量明显减少,而过表达Hp可明显减少RSL3引起的细胞的死亡(图4C)。而且,Hp可显著抑制由RSL3诱导引起的PTGS2表达增加和GPX4表达下调(图4D、E)。利用C11-BODIPY581/591荧光探针标记细胞内脂质过氧化,给予RSL3刺激后,流式检测和免疫荧光结果显示C11-BODIPY581/591染色阳性的AML-12肝细胞明显增多,而Hp过表达后C11-BODIPY581/591荧光染色明显减少,提示Hp过表达可以显著抑制RSL3诱导的脂质过氧化反应(图4F、G)。以上结果表明,Hp可以抑制RSL3诱导的肝细胞过氧化反应和铁死亡。
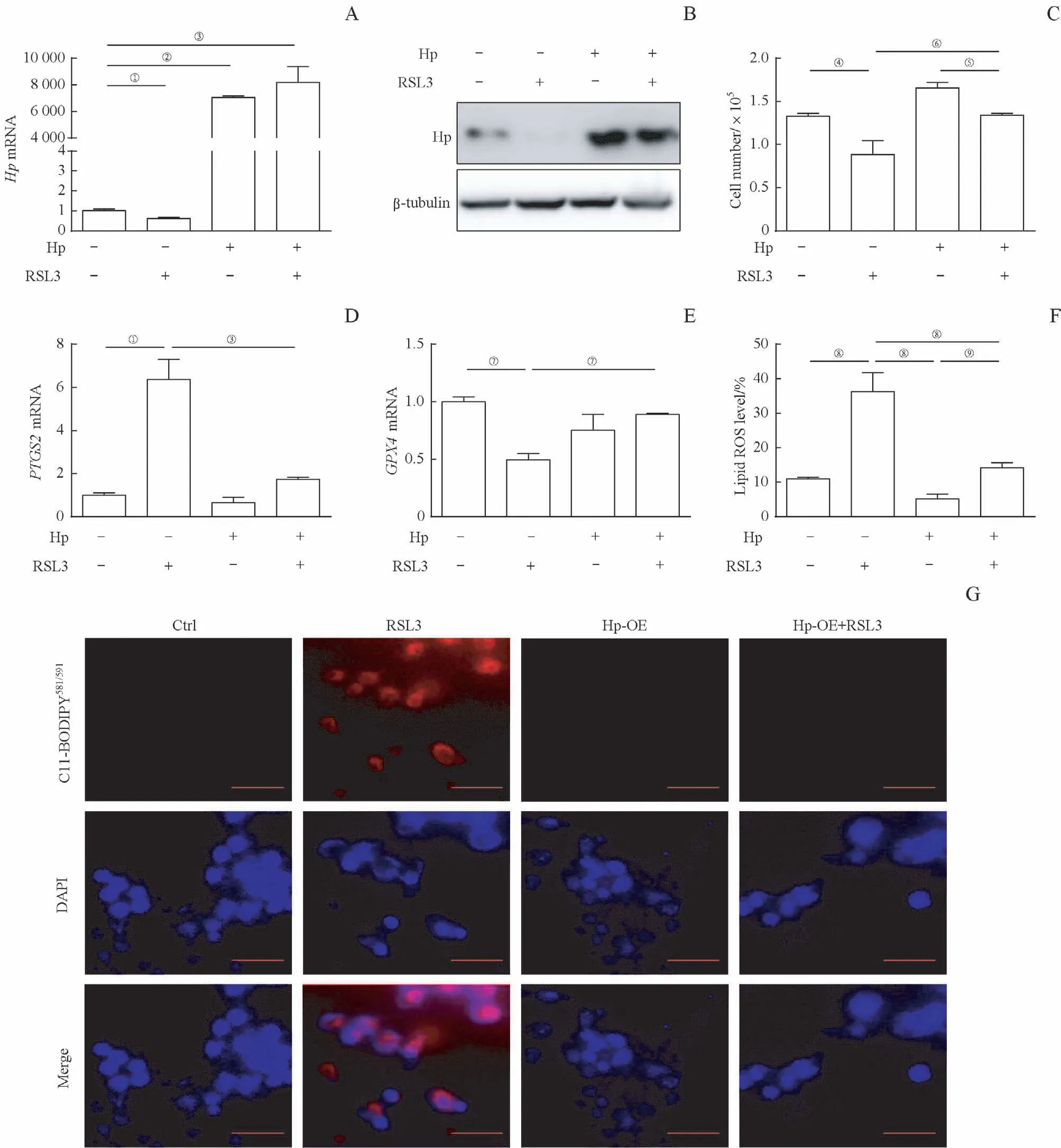
图4 Hp过表达抑制RSL 3诱导的肝细胞铁死亡Fig 4 Hp overexpression inhibiting hepatocyteferroptosis induced by RSL3
2.5 Hp通过抑制ERK1/2磷酸化减轻肝细胞铁死亡
ERK通路的激活与铁死亡密切相关[14],因此,为了进一步揭示Hp抑制肝细胞铁死亡的分子机制,我们首先利用Western blotting检测了ERK1/2蛋白磷酸化水平,发现给予RSL3刺激后,AML-12肝细胞中ERK1/2蛋白的磷酸化增强,而Hp过表达可以明显抑制RSL3引起的ERK1/2蛋白磷酸化(图5A)。而且,Hp敲低可明显加重RSL3诱导的AML-12肝细胞死亡(图5B、C),PTGS2和ACSL4mRNA表达的增加(图5D、E),GPX4mRNA表达的降低(图5F),以及脂质过氧化反应的增强(图5G、H),提示Hp敲低可明显加重肝细胞铁死亡。而ERK1/2抑制剂SCH772984可以明显减轻Hp敲减后对脂质过氧化和铁死亡的加重(图5C~H)。以上结果表明,Hp主要通过抑制ERK1/2磷酸化减轻肝细胞铁死亡。
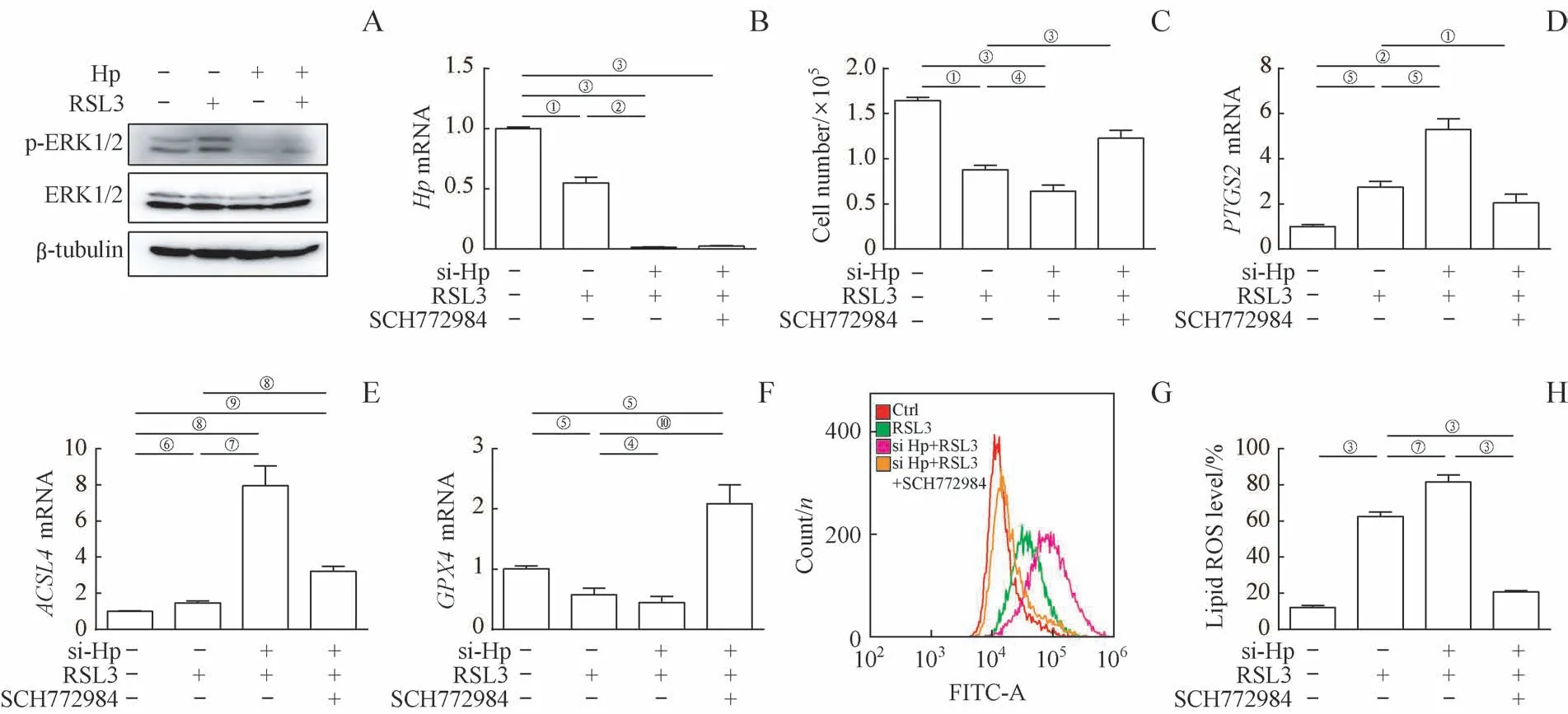
图5 Hp通过抑制ERK1/2磷酸化减轻肝细胞铁死亡Fig 5 Hp suppressed hepatocyte ferroptosis by inhibiting ERK1/2 phosphorylation
3 讨论
细胞铁死亡主要通过Fe2+和脂质过氧化产物累积对细胞造成损伤和死亡。Fe2+作为含铁酶的辅助因子在氧化应激反应、能量代谢和铁硫蛋白产生等代谢生化过程中均发挥重要作用[1,18-19]。细胞内Fe2+增多会诱导细胞发生铁死亡[1,20-22]。在本研究中,我们利用高铁饮食饲喂构建了小鼠肝脏铁死亡的模型,通过肝脏H-E染色(图1C),血清GPT(图1E)和GOT检测(图1F)结果可以发现高铁饮食可以明显诱导小鼠的肝脏损伤。同时,普鲁士蓝染色(图1G)和肝脏铁含量检测(图1H)证实了高铁饮食可以引起肝细胞铁沉积。4-HNE和MDA的检测证实了高铁饮食导致了脂质过氧化产物的累积(图1I、J)。而realtime PCR结果显示,铁死亡过程中重要的分子PTGS2[3,12]、ACSL4[23-24]和GPX4[3-4]发生了相应的变化(图1K~M),提示高铁饮食诱导的肝细胞死亡形式主要为铁死亡,这些结果与文献报道一致[12,25]。同时RNA-seq结果分析也表明,氧化还原反应和细胞死亡通路被明显激活,这些结果均进一步证明高铁饮食可以促进肝细胞的铁死亡进而引起肝损伤和纤维化,高铁饮食小鼠模型是一种比较理想的研究肝细胞铁死亡机制的动物模型。
利用RNA-seq结果进行GO和GSEA分析发现,急性时相蛋白Hp参与了高铁饮食调控的铁死亡相关信号通路,而且实验证实Hp在HID组小鼠肝脏及RSL3诱导的肝细胞中表达水平明显下调,提示Hp可能参与了铁死亡发生过程。为了进一步验证Hp在肝细胞铁死亡中的功能,我们给予Hp过表达的AML-12肝细胞铁死亡诱导剂RSL13,利用细胞计数、real-time PCR和C11-BODIPY581/591免疫荧光及流式分析分别检测了细胞的死亡、铁死亡相关基因(PTGS2、ACSL4、GPX4)以及肝细胞的脂质氧化水平,发现Hp过表达可以明显减轻RSL3引起的AML-12肝细胞的死亡和脂质过氧化产物累积(图4)。而相反的,Hp敲低则可明显加重RSL3引起的肝细胞铁死亡(图5)。由此,我们证实Hp是肝细胞铁死亡的新的抑制分子。而有文献报道,铁死亡诱导剂erastin或RSL3可上调肺癌细胞中Hp的表达,敲除Hp后可以减轻RSL3引起的肺癌细胞铁死亡并增加肺癌细胞对erastin或者RSL3的耐药性[15]。这与我们结果存在的差异可能是由于我们使用的是正常的肝细胞,而正常体细胞与肿瘤细胞对于铁死亡的敏感性和机制不同[26-28]。Hp在肝癌细胞铁死亡中发挥什么作用还有待于进一步研究。
ERK1/2是丝氨酸/苏氨酸蛋白激酶,属于丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)家族,广泛存在于真核细胞中。ERK1/2在细胞表面受体到细胞核的信号转导中起关键作用,激活的p-ERK1/2磷酸化细胞质或细胞核内的底物,从而诱导特定蛋白的表达或激活,导致细胞增殖、分化、凋亡等过程的调控[29-30]。而且,ERK信号通路异常参与多种肝脏疾病的发生发展。有研究[31]表明,在ALD小鼠模型中,肝细胞ERK1/2的活性受到抑制;但是也有文献[32-33]指出,慢性酒精饮食可以增强库普弗细胞中ERK1/2激活的反应,促进肿瘤坏死因子合成增加和炎症反应增强。这可能是由于研究的细胞和酒精处理时间不同[34],但以上研究结果提示ERK1/2信号通路在ALD中是失调的。而在NAFLD中,小鼠肝细胞特异性敲除IL-11受体亚基α-1(interleukin 11 receptor,alpha chain 1,IL11ra1)可以减少IL-11诱导的肝细胞ROS产生,以及ERK和JNK通路激活,从而减轻高脂高糖饮食诱导的肝细胞损伤和NASH[35]。同时,ERK通路激活后通过负调控TGF-β/Smad信号,促进成纤维细胞的增殖和分化,上调胶原基因的表达和合成以及细胞外基质沉积,进而促进肝纤维化的进展[30]。由此,ERK1/2通路在铁死亡相关的肝病(ALD、NAFLD和肝纤维化)[7]的发生与发展中均发挥重要作用,但ERK1/2通路是否可以调控肝细胞铁死亡以及是否通过调控铁死亡参与肝病的发生发展尚不清楚。而目前有文献[14]报道在大鼠肾上腺嗜铬细胞瘤细胞株(PC12细胞)铁死亡发生过程中,ERK通路被激活,ERK1/2磷酸化水平上调。我们的结果表明,高铁饮食和RSL3可以明显上调肝细胞ERK1/2磷酸化水平,而Hp过表达后ERK通路得到抑制,提示ERK1/2信号通路可能介导了Hp抑制肝细胞铁死亡的功能。随后的实验进一步证实,ERK1/2抑制剂SCH772984可以明显减轻Hp敲减导致的铁死亡的加重,提示Hp是通过抑制ERK1/2活性抑制肝细胞铁死亡,可能在铁死亡相关的肝病中发挥重要的作用。
总之,本研究表明Hp是一种新的肝细胞铁死亡抑制因子,可以通过抑制ERK1/2信号通路活化来抑制肝细胞铁死亡。这为治疗铁死亡引起的肝损伤或利用铁死亡治疗肝癌提供了新的靶点和证据。
参·考·文·献
[1] Tang D,Chen X,Kang R,et al.Ferroptosis:molecular mechanisms and health implications[J].Cell Res,2021,31(2):107-125.
[2] Liu J,Song X,Kuang F,et al.NUPR1 is a critical repressor of ferroptosis[J].Nat Commun,2021,12(1):647.
[3] Yang WS,SriRamaratnam R,Welsch ME,et al.Regulation of ferroptotic cancer cell death by GPX4[J].Cell,2014,156(1-2):317-331.
[4] Dixon SJ,Lemberg KM,Lamprecht MR,et al.Ferroptosis:an irondependent form of nonapoptotic cell death[J].Cell,2012,149(5):1060-1072.
[5] Kagan VE,Mao G,Qu F,et al.Oxidized arachidonic and adrenic PEs navigate cellsto ferroptosis[J].Nat Chem Biol,2017,13(1):81-90.
[6] Yuan H,Li X,Zhang X,et al.Identification of ACSL4 as a biomarker and contributor of ferroptosis[J].Biochem Biophys Res Commun,2016,478(3):1338-1343.
[7] Macías-Rodríguez RU, Inzaugarat ME, Ruiz-Margáin A, et al.Reclassifying hepatic cell death during liver damage:ferroptosis-A novel form of non-apoptotic cell death?[J].Int JMol Sci,2020,21(5):1651.
[8] Gao W,Zhao J,Gao Z,et al.Synergistic interaction of light alcohol administration in the presence of mild iron overload in a mouse model of liver injury:involvement of triosephosphate isomerase nitration and inactivation[J].PLoSOne,2017,12(1):e0170350.
[9] Liu CY,Wang M,Yu HM,et al.Ferroptosis is involved in alcohol-induced cell deathin vivoandin vitro[J].Biosci Biotechnol Biochem,2020,84(8):1621-1628.
[10] Tsurusaki S,Tsuchiya Y,Koumura T,et al.Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis[J].Cell Death Dis,2019,10(6):449.
[11] 贺明,魏倩,张颖婷.铁死亡相关机制与其在肝脏相关疾病中作用的研究进展[J].上海交通大学学报(医学版),2020,40(11):1519-1523.
[12] Wang H,An P,Xie E,et al.Characterization of ferroptosis in murinemodels of hemochromatosis[J].Hepatology,2017,66(2):449-465.
[13] Fang X,Wang H,Han D,et al.Ferroptosis as a target for protection against cardiomyopathy[J].Proc Natl Acad Sci USA,2019,116(7):2672-2680.
[14] Wu C,Zhao W,Yu J,et al.Induction of ferroptosis and mitochondrial dysfunction by oxidativestressin PC12 cells[J].Sci Rep,2018,8(1):574.
[15] Li G,Yang J,Zhao G,et al.Dysregulation of ferroptosis may involve in the development of non-small-cell lung cancer in Xuanwei area[J].J Cell Mol Med,2021,25(6):2872-2884.
[16] di Masi A,De Simone G,Ciaccio C,et al.Haptoglobin:from hemoglobin scavenging to human health[J].Mol Aspects Med,2020,73:100851.
[17] Shu H,Zhang S,Kang X,et al.Protein expression and fucosylated glycans of the serum haptoglobin-βsubunit in hepatitis B virus-based liver diseases[J].Acta Biochim Biophys Sin(Shanghai),2011,43(7):528-534.
[18] Outten FW,Theil EC.Iron-based redox switches in biology[J].Antioxid Redox Signal,2009,11(5):1029-1046.
[19] Wang Y,Liu Y,Liu J,et al.NEDD4L-mediated LTF protein degradation limits ferroptosis[J]. Biochem Biophys Res Commun,2020,531(4):581-587.
[20] Yuan H,Li X,Zhang X,et al.CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation[J]. Biochem Biophys Res Commun,2016,478(2):838-844.
[21] Alvarez SW,Sviderskiy VO,Terzi EM,et al.NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis[J].Nature,2017,551(7682):639-643.
[22] Gao M,Monian P,Pan Q,et al.Ferroptosis is an autophagic cell death process[J].Cell Res,2016,26(9):1021-1032.
[23] Doll S,Proneth B,Tyurina YY,et al.ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition[J].Nat Chem Biol,2017,13(1):91-98.
[24] Lei G,Zhang Y,Koppula P,et al.The role of ferroptosisin ionizing radiationinduced cell death and tumor suppression[J].Cell Res,2020,30(2):146-162.
[25] Yu Y,Jiang L,Wang H,et al.Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis[J].Blood,2020,136(6):726-739.
[26] Mou Y,Wang J,Wu J,et al.Ferroptosis,anew form of cell death:opportunities and challengesin cancer[J].JHematol Oncol,2019,12(1):34.
[27] Gnanapradeepan K,Basu S,Barnoud T,et al.The p53 tumor suppressor in the control of metabolism and ferroptosis[J].Front Endocrinol(Lausanne),2018,9:124.
[28] Qiu Y,Cao Y,Cao W,et al.The application of ferroptosis in diseases[J].Pharmacol Res,2020,159:104919.
[29] Sreekanth GP,Chuncharunee A,Sirimontaporn A,et al.Role of ERK1/2 signaling in dengue virus-induced liver injury[J].Virus Res,2014,188:15-26.
[30] Long T,Wang L,Yang Y,et al.Protective effects of trans-2,3,5,4'-tetrahydroxystilbene 2-O-β-D-glucopyranoside on liver fibrosis and renal injury induced by CCl4viadown-regulating p-ERK1/2 and p-Smad1/2[J].Food Funct,2019,10(8):5115-5123.
[31] Sampey BP,Stewart BJ,Petersen DR.Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activityvia4-hydroxynonenal[J].JBiol Chem,2007,282(3):1925-1937.
[32] Thakur V,Pritchard MT,McMullen MR,et al.Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells:role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-αproduction[J].JLeukoc Biol,2006,79(6):1348-1356.
[33] Mandrekar P,Szabo G.Signalling pathways in alcohol-induced liver inflammation[J].JHepatol,2009,50(6):1258-1266.
[34]Wang Z,Yao T,Song Z.Involvement and mechanism of DGAT2 upregulation in the pathogenesis of alcoholic fatty liver disease[J].J Lipid Res,2010,51(11):3158-3165.
[35] Dong J,Viswanathan S,Adami E,et al.Hepatocyte-specific IL11 cissignaling drives lipotoxicity and underlies the transition from NAFLD to NASH[J].Nat Commun,2021,12(1):66.
猜你喜欢
中国油脂(2022年5期)2022-05-31
中国典型病例大全(2022年13期)2022-05-10
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
现代临床医学(2021年6期)2021-11-20
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年1期)2021-02-07
食品界(2018年8期)2018-09-03
饮食科学(2016年12期)2017-01-07
食品工业科技(2014年13期)2014-03-11
