
MaR1治疗脂多糖诱导的急性肺损伤小鼠的效果和机制*
2021-09-07 02:34王强林飞胡召锟林锦源黄冰潘灵辉
广东医学 2021年8期
王强, 林飞, 胡召锟, 林锦源, 黄冰, 潘灵辉
广西医科大学附属肿瘤医院麻醉科(广西南宁 530021)
急性肺损伤(ALI)是临床上急性呼吸衰竭的重要病因,可由感染性和非感染性等方面因素引起,ALI可导致肺泡上皮、血管内皮细胞损伤和肺血管通透性增加,它的特征是嗜中性粒细胞大量涌入,肺泡-毛细血管屏障损伤导致间质性水肿和呼吸功能受损[1-2]。ALI的危险因素众多,ALI动物模型是研究 ALI 发病机制和探索其治疗方法的重要手段。虽然没有任何一个动物模型能完全复制人类ALI的所有特征,但脂多糖(LPS)诱导ALI被认为是理想的动物模型之一,能够较好复制ALI的发病机制和预后[3-4]。来自革兰阴性细菌外膜的LPS在ALI实验动物模型中通过产生活性氧(ROS)和炎症反应来增强氧化应激,氧化应激在LPS诱导的ALI的发病机制中也起着重要作用[5-6]。目前ALI的治疗尚无特效药,一般是找出病因,然后对原发病进行针对性治疗, ALI的常规治疗药物和方法主要包括机械通气疗法、舒血管药、表面活性剂、抗氧化剂、糖皮质激素、抗炎药等。最近,许多学者开始研究具有抗氧化或抗炎特性的Maresin以及化学异构体,发现Maresin-1(MaR1)在体内、离体实验能够抑制中性粒细胞的浸润、促进巨噬细胞的趋化和表型转化并增强其吞噬功能、刺激组织再生、控制疼痛,从多个环节减轻炎症反应强度,促进炎症消退,有望成为调控炎症反应的治疗靶点[7]。但有关MaR1在ALI模型上抗炎效果和作用机制鲜有报道研究。因此,本研究收集自2018年1月至2019年8月MaR1治疗小鼠ALI模型的实验数据,以探讨MaR1治疗LPS诱导的ALI小鼠模型中炎症的效果以及其潜在的机制。
1 材料与方法
1.1 ALI模型制备 雄性SPF级BALB/c小鼠45只,体重20~25 g,由广西医科大学动物实验中心提供。术前禁食12 h,自由饮水。2%戊巴比妥钠(80 mg/kg)腹腔注射麻醉小鼠。通过气管内滴注LPS(3 mg/kg)诱导小鼠ALI模型。实验动物平均随机分为3组。对照组:小鼠经口气管插管滴入生理盐水,1 h后尾静脉注射生理盐水。LPS组:小鼠经口气管插管滴入LPS(3 mg/kg),1 h后尾静脉注射生理盐水。MaR1组:小鼠经口气管插管滴入LPS(3 mg/kg),1 h后尾静脉注射MaR1(1 ng/只)。所有小鼠在气管给予LPS 24 h后处死。
1.2 小鼠肺组织病理学 处死小鼠解剖肺组织,并固定在10%中性甲醛缓冲溶液中。将固定的组织包埋在石蜡中,切成4 μm的厚度,去石蜡,然后重新水化。肺损伤程度通过HE染色进行评估。对于每个载玻片,每张载玻片都用光学显微镜以全盲方式手动检查,所有病变均以完全不可见的方式使用10倍和20倍物镜。主要的组织学损害包括白细胞浸润和肺泡壁增厚,分为以下等级:0分=无损害,1分=轻度,2分=适中,3分=严重。
1.3 免疫印迹 在含有蛋白酶和磷酸酶抑制剂混合物(Thermo Scientific)的T-PER组织蛋白提取试剂(Thermo Scientific,美国)中将肺组织匀浆(1/10,W/V)。使用Bradford试剂时确定每个样品的蛋白质浓度。通过4%~12%SDS-聚丙烯酰胺凝胶电泳分离等量的总蛋白(30 μg),然后转移到聚二氟乙烯膜上,将膜与封闭液孵育,然后与以下一级抗体和稀释液在4℃过夜孵育:Erk,p-Erk,JNK,p-JNK,p38MAPK,p-p38MAPK,p65NF-κB,p-p65NF-κB,β-肌动蛋白(1∶1 000稀释),诱导型一氧化氮合酶(iNOS),Nrf2,HO-1,NQO1和TNF-α。将膜用含0.05%Tween 20(TBST)的Tris缓冲盐水洗涤3次,然后与1∶10 000稀释的辣根过氧化物酶偶联的二抗孵育1 h在室温下。再次用TBST将印迹洗涤3次。使用增强型化学发光试剂盒形成蛋白条带,一式两份测量每个样品的蛋白质表达。使用化学发光扫描仪(LI-COR,美国)确定每个蛋白条带的光密度分析。
1.4 氧化应激标记分析 收集的上清液用于测定氧化应激水平,通过测定脂质过氧化的标志物硫代巴比妥酸反应性物(TBARS)和谷胱甘肽(GSH)的含量来进行。将200 μL组织裂解液与500 μL三氯乙酸混合,并在10 min内孵育。在4℃下测定TBARS。孵育后,将样品以3 000×g离心10 min。在4℃下。收集上清液并与0.025 mol/L TBA混合并温育45 min。在65℃下。使用酶标仪在540 nm处测定吸光度,并使用标准曲线(范围0~20 μmol/L)将其转换为μmol。样品中的TBASRS水平表示为nmol/mg蛋白。将50 μL肺组织裂解液与350 μL 5%偏磷酸混合,然后涡旋20 s。确定谷胱甘肽浓度。将样品以3 000×g离心10 min。在4℃下。将上清液与作为显色剂的5,5′-二硫代双-2-硝基苯甲酸(DTNB)混合,并添加谷胱甘肽还原酶,并孵育5 min。在室温下孵育后,将烟酰胺腺嘌呤二核苷酸磷酸(NADPH)加入样品中,并使用酶标仪在415 nm下测定吸光度。通过在标准曲线上外推计算GSH的含量,并表达nmol/mg蛋白。称重的肺组织用玻璃珠和冰冷的PBS(pH=7.4)研磨,得到1∶9(W/V)的完整匀浆。将匀浆以11 000×g离心15 min。在4℃时丢弃细胞碎片。
1.5 统计学方法 使用SPSS 22.0统计软件,3组之间差异采用方差分析(ANOVA)检验,然后使用LDS的多重比较检验进行多重比较检验。以P<0.05为差异有统计学意义。
2 结果
2.1 各组小鼠肺组织损伤评分比较 对照组肺组织损伤评分0分,LPS组肺组织损伤评分(2.81±1.35)分,MaR1组肺组织损伤评分(1.24±0.73)分。MaR1组与LPS组肺组织评分之间差异有统计学意义(t=3.96,P=0.000)。经LPS诱导ALI的小鼠表现出明显的组织病理学变化,包括肺泡壁增厚,支气管周围和血管周围白细胞浸润浸润。用MaR1治疗可减少炎症细胞的浸润并减少肺组织中肺泡壁的增厚,见图1。
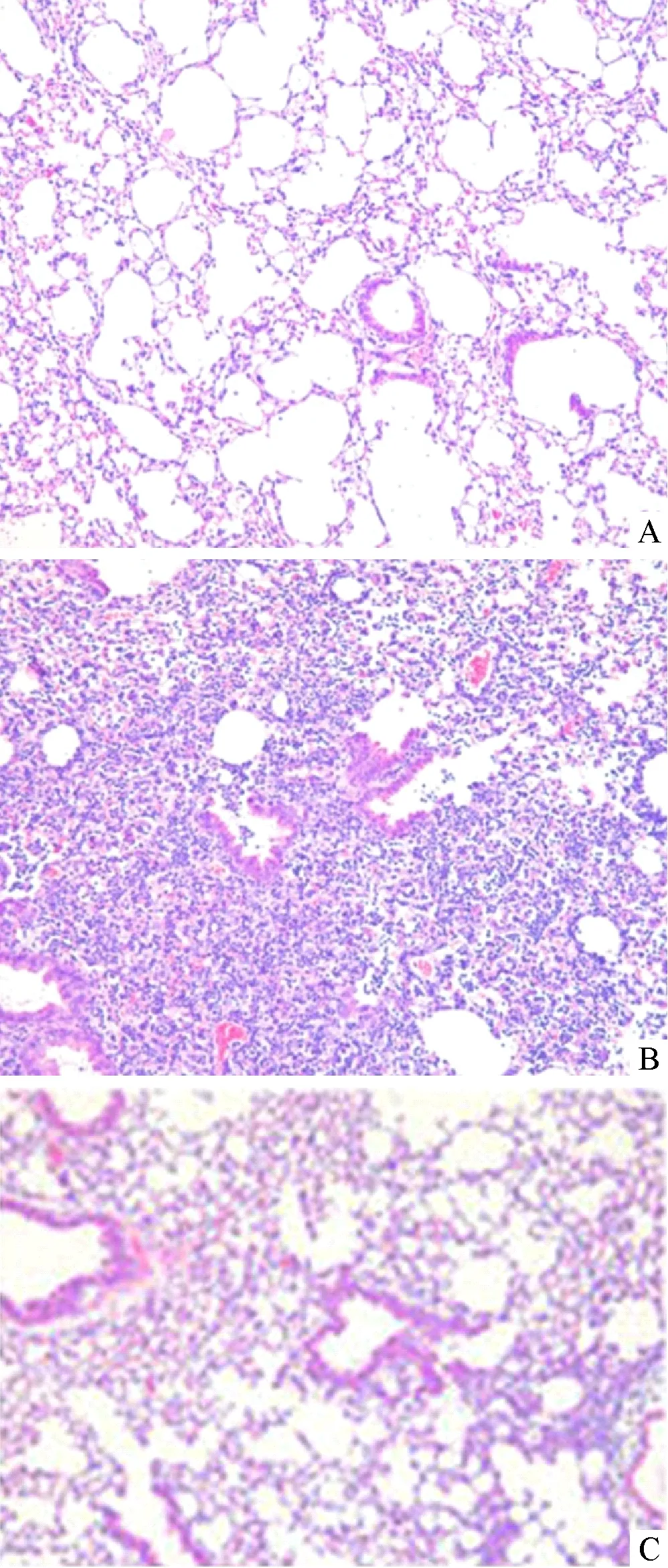
注:A:对照组小鼠肺组织未见损伤表现;B:LPS组小鼠肺泡壁增厚,支气管周围和血管周围白细胞浸润浸润;C:MaR1组小鼠肺泡壁增厚,支气管周围和血管周围白细胞浸润浸润治疗后较前好转
2.2 各组小鼠肺组织MAPKs、NF-κB和炎症介质活化比较 与对照组比较,LPS组肺组织MAPKs蛋白(JNK、Erk和p38MAPK)表达不同程度上调(P<0.05),与LPS组比较,MaR1组治疗小鼠肺组织中JNK、Erk、p65NF-κB和p38MAPK的磷酸化显著下降(P<0.05)。与对照组比较,LPS组肺组织NF-κB蛋白表达上调(P<0.05)。与LPS组比较,MaR1组治疗小鼠肺组织中NF-κB蛋白表达下调(P<0.05)。与LPS组比较,MaR1治疗小鼠的肺组织中iNOS和TNF-α表达水平显著减低(P<0.05),见图2和表1。
表1 3组ALI小鼠MAPK、p65NF-κB和炎症介质活化结果对比
2.3 各组小鼠肺组织Nrf2、抗氧化酶表达和TBARS GSH比较 与对照组比较,LPS组Nrf2、抗氧化酶表达和TBARS GSH之间差异有统计学意义(P<0.05),与LPS组比较,MaR1组小鼠在肺组织中的Nrf2核易位显著增加(P<0.05),MaR1组肺组织HO-1和NQO1的表达水平增加(P<0.05),MaR1组小鼠显示出TBARS水平的显著升高和GSH含量的显著降低(P<0.05),见图3和表2。
表2 3组ALI小鼠肺组织Nrf2表达、抗氧化酶以及TBARS GSH结果对比
3 讨论
革兰阴性细菌感染居ALI高危因素的首位,LPS是革兰阴性细菌细胞壁的关键成分,也是其致病成分,能诱发巨噬细胞活化和释放炎症介质。当其暴露于动物和人类中时,LPS导致巨噬细胞和淋巴细胞分泌和释放的一系列炎症因子(如TNF、IL-1、IL-6),显示出微血管肺损伤的主要特征,包括肺组织中的白细胞积聚、肺水肿、严重的肺部炎症和病死率[8-9]。LPS诱导的动物模型可重现肺部上皮和内皮屏障的急性损伤以及气隙中的急性炎症反应。因此,LPS诱导的损伤是理想的体内实验模型,非常类似于ALI/ARDS。LPS诱导ALI给药途径包括鼻内、气管内、腹膜内或静脉内或通过LPS两次打击方法(即小鼠腹膜内注射小剂量的LPS,然后气管滴注中剂量的LPS)等,有学者认为LPS两次打击模型似乎更稳定和可靠,更接近于人类ARDS的诊断标准,但也有学者研究表明LPS在鼻内或气管内给药后24 h对小鼠造成ALI,而静脉内和腹膜内给药并未导致组织特异性或类似程度的肺损伤[10-11]。
本研究是小鼠经口气管插管滴入LPS,LPS暴露促进炎性细胞因子的释放,诱导ROS产生氧化应激,造成肺组织直接损失,建立了ALI模型。尽管肺损伤的机制因病因而异,血管通透性增加,细胞因子的过度产生,白细胞集聚和表面活性剂功能障碍等上皮和内皮细胞的损害,从而导致间质和肺泡肺水肿,肺泡塌陷和低氧血症,但当前没有针对ARDS的特定有效疗法。特异性促炎症消退介质是一系列非不饱和脂肪酸合成的脂质介质,它们具有抗炎和促炎症消退的作用,主要包括脂氧素(lipoxins,LXs)、消退素(resolvins,Rvs)、保护素和Maresins等,其中,Maresins是由巨噬细胞合成的二十二碳六烯酸衍生的促炎症消退介质家族[7]。在炎症刺激下,二十二碳六烯酸在14-脂肪氧化酶催化下合成中间代谢产物14S- HpDHA,进一步在13/14-环氧化酶的作用下缩合生成Maresin。MaR1是最早发现的Maresin的化学异构体,能够抑制中性粒细胞的浸润、促进巨噬细胞的趋化和表型转化并增强其吞噬功能、刺激组织再生、控制疼痛,从多个环节减轻炎症反应强度,促进炎症消退。

注:3组小鼠肺组织的MAPKs、p65NF-κB、iNOS和TNF-α的蛋白水平表达情况,β-肌动蛋白内参抗体
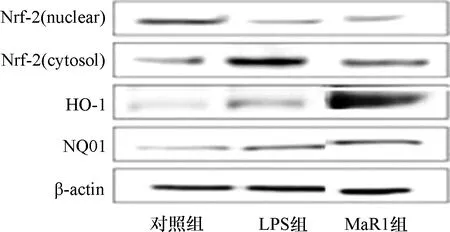
注:3组小鼠组织的Nrf-2、HO-1和NQO1的蛋白质水平表达情况,β-肌动蛋白内参抗体
本研究发现MaR1治疗LPS诱导的ALI模型能够抑制p65NF-κB的磷酸化并减少促炎介质,表明MaR1可能通过抑制NF-κB活化来抑制LPS诱导的肺部炎症反应,从而导致炎症介质或细胞因子的抑制。因为LPS可以通过与TLR4结合来激活免疫细胞,从而触发TRIF或MyD88依赖性途径。这些途径调节炎症反应,从而促进MAPK和NF-κB的激活。在MAPK家族中,JNK、Erk和p38MAPK等一系列蛋白激酶参与多种细胞反应,包括炎症相关途径[12-13]。这些MAPKs被细胞外或细胞内信号激活,继而促进炎症过程中早期促炎性细胞因子的产生,有学者研究表明LPS诱导的ALI模型中MAPK信号通路被激活,与本研究结果类似。此外,本研究发现MaR1治疗可有效抑制LPS诱导的ALI小鼠模型中JNK、Erk和p38MAPK的磷酸化,表明MAR1治疗通过阻断MAPK的活化有效地抑制了炎症反应。NF-κB是各种炎症介质/细胞因子的上游调节剂,在调节TLR4介导的炎症反应中起着重要作用。在NF-κB信号通路中,IκB激酶可以使IκB磷酸化,然后将p65-p50二聚体转运到细胞核中,并与DNA结合以调节iNOS和TNF-α等促炎性基因的表达[14-15]。LPS可以通过激活NF-κB信号传导来诱导促炎性细胞因子的释放,MaR1治疗能抑制NF-κB可以减弱LPS诱导的ALI。
本研究发现MaR1治疗后肺组织中HO-1和NQO1的表达水平高于LPS诱导的ALI模型小鼠,表明MaR1可能通过HO-1和NQO1的上调而促进Nrf2的核易位。氧化应激在ALI的发病机制中起着重要的作用,氧化应激特征是ROS的积累促进促炎细胞因子的表达和促炎细胞的浸润而加剧了炎症反应,从而导致ALI[16-18]。MaR1有效减弱了脂质过氧化作用,表明MaR1具有强效的抗氧化特性。 Nrf2在许多与炎症和氧化应激相关的疾病中起着至关重要的作用,其通过ARE介导的HO-1和NQO1等多种抗氧化酶保护ROS免受各种组织和细胞的损伤[19-20]。在氧化应激条件下,Nrf2从胞质溶胶转移到细胞核,随后与ARE结合,从而导致HO-1和NQO1的表达。它提供了抗氧化应激的宿主防御机制,并有助于抗炎活性,增强Nrf2激活可以减弱LPS诱导的ALI[21]。故认为MAR1对LPS诱导ALI的氧化应激的保护作用机制可能是通过Nrf2的激活及其有效的抗氧化活性相关。
综上所述,MaR1治疗能有效抑制MAPK和p65NF-κB的磷酸化,达到ALI模型小鼠炎性细胞向肺组织的浸润,Nrf2通过ARE介导的HO-1和NQO1等多种抗氧化酶保护ROS免受各种组织和细胞的损伤。MaR1治疗对炎症内源性调控的作用, 它将有望成为ALI调控炎症反应的治疗靶点。
猜你喜欢
现代临床医学(2021年4期)2021-07-31
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际呼吸杂志(2019年8期)2019-04-29
国际呼吸杂志(2019年8期)2019-04-29
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
老年医学与保健(2017年6期)2017-02-06
西南军医(2016年6期)2016-01-23
华南农业大学学报(2015年5期)2015-12-04
癌变·畸变·突变(2015年3期)2015-02-27
