
DOT1L对骨肉瘤MG63细胞生物学行为的影响及其作用机制*
2021-09-07 02:37王洪伸林涌鹏尹萌辰常君丽陈博来
广东医学 2021年8期
王洪伸, 林涌鹏, 尹萌辰, 常君丽, 陈博来△
1广州中医药大学第二附属医院骨伤一科(广东广州 510120); 上海中医药大学附属龙华医院 2骨伤科, 3上海中医药大学脊柱病研究所(上海 200032)
骨肉瘤是起源于骨间叶细胞的原发恶性骨肿瘤,约占恶性骨肿瘤的20%,其发病年龄呈双峰状,75%发生于儿童和青年为原发性;25%发生于中老年为继发性[1-3]。首次确诊后转移患者5年生存率低于30%,具有极高危害性[4]。因此寻找有效的药物治疗靶点临床迫切需要,而DNA损伤与修复途径一直是关注靶点。骨肉瘤DNA损伤后无法及时修复,将逐步导致细胞增殖和分化失控,诱发恶性增殖[4-5];而DNA损伤修复功能过强,将导致细胞修复加速,增加肿瘤发生率或耐药性和抵抗力[5]。DOT1L催化H3K79甲基化促进DNA损伤修复、端粒酶沉默[6]。有肿瘤细胞测序报道和CBioPortal肿瘤数据库表明,DOT1L在部分肉瘤中呈高表达[7];而SIRT1在骨肉瘤中呈现促癌基因高表达特性[8-9],参与DNA双链损伤修复[8],能够与DOT1L相互作用,增强H3K79甲基化的分布和活性。因此推测DOT1L可能通过DOT1L/H3K79me2-SIRT1及下游途径调控骨肉瘤细胞增殖、凋亡、转移等生物学行为。为验证上述科学假设,2019年1月至2020年9月,本研究首次采用shRNA敲减骨肉瘤细胞DOT1L基因,观察MG63细胞增殖、迁移、DNA损伤的影响,探讨DOT1L成为骨肉瘤治疗新靶点的具体机制与潜能。
1 材料与方法
1.1 主要材料 人骨肉瘤细胞株MG63购自上海中科院细胞库;MEM培养液和胎牛血清购自美国Gibco公司,DOT1L抗体(ab64077)、XPA抗体(ab85914)、XPC抗体(ab155025)、H3K79me2(ab3594)、Histone H3抗体(ab1791)、β-actin抗体 (ab8227)均购自美国Abcam公司,SIRT1抗体(#8469)购自美国Cell Signaling Technology公司。hDOT1L shRNA TRC系列质粒购自美国GE Healthcare Dharmacon公司。RIPA裂解液、SDS缓冲液、SDS-PAGE凝胶配制试剂盒、BCA蛋白浓度测定试剂盒、BeyoECL Plus试剂盒、TUNEL试剂盒、DAPI等均购自上海碧云天公司,RNA提取试剂盒和逆转录cDNA试剂盒均购自Invitrogen公司;EMLBSLR显微镜购自日本Leica公司、BH-20光学显微镜购自日本Olympus公司。酶标仪、细胞培养箱来自美国赛默飞科技公司、CKX31普通光学显微来自日本奥林巴斯公司,细胞培养板购自美国Corning公司,双垂直电泳仪、转印电泳仪、凝胶成像系统均购自美国Bio-Rad公司。
1.2 细胞培养与转染 MG63细胞用含10%胎牛血清的MEM 培养基,于37℃、5% CO2恒温培养箱中培养,每天更换新鲜培养液,待细胞生长融合至70%~80%进行传代。按照LipofectamineTM2000转染试剂盒说明书的方法,将hDOT1L-shRNA及其阴性对照(shRNA-Scramble)转染至MG63细胞,定义为hDOT1L-shRNA组和Scramble组;培养4~6 h后更换新鲜完全培养基,继续培养24 h后,嘌呤霉素筛选收集各组细胞进行后续实验。
1.3 实时荧光定量PCR检测MG63细胞中DOT1L mRNA的表达 用RNA提取试剂盒分别提取各组细胞中的总RNA,将提取的总RNA按照逆转录试剂盒说明书的操作步骤逆转录为cDNA,然后用RealtimePCR检测各组细胞中DOT1L水平。PCR反应体系:ddH2O 7.2 μL,SybrGreen qPCR Master Mix(2×)10.0 μL,10 μmol/L前后引物各0.4 μL,cDNA 2.0 μL,终体积为20 μL。扩增条件:94℃预变性10 min(94℃变性20 s,55℃退火20 s,72℃延伸20 s)40个循环。DOT1L PCR引物序列:正向引物为:5′-GAAGAGCAGTGAGAAGGGC-3′,反向引物为:5′-TTTTCAAGTGTGGACGAGG-3′,GAPDH 正向引物为:5′-AGAAGGCTGGGGCTCATTTG-3′,反向引物为:5′-AGGGGCCATCCACAGTCTTC-3′,采用2-△△CT法分析内参对照组和目的基因组之间的表达差异。实验重复3次。
1.4 MTT法及检测MG63细胞的增殖能力 转染完成后调整细胞浓度为5×104·mL-1,接种至96孔培养板(100 μL/孔),37℃、5% CO2培养箱内培养,分别在24、48、72 h后在避光环境下每孔加入5 mg/mL MTT(10 μL/孔),37℃、5%CO2培养箱内培养避光孵育3~4 h,弃去上清液,加入200 μL DMSO,室温于摇床振荡10 min,用酶标仪492 nm波长测出同一时间点OD值,用测得的OD值进行细胞增殖影响的分析。
1.5 划痕实验检测MG63细胞的迁移能力 取3.5 cm直径培养皿背侧均匀划横线,间隔0.5~1 cm,横穿过孔,每孔至少穿过5条线。取对数生长期MG63细胞,调整细胞浓度为3×105·mL-1,接种于3.5 cm培养皿(2 mL/皿),常规培养24 h后采用Lipofectamine 2000制备转染复合物,转染成功后继续培养6 h,弃去上清液,200 μL枪头垂直于背侧横线划痕,PBS清洗脱落细胞并弃除上清液。继续培养48 h,分别于倒置显微镜下记录0、48 h的细胞迁移,根据距离计算细胞迁移率。实验重复3次。
1.6 TUNEL法检测细胞DNA损伤 收集对数生长期MG63细胞,调整细胞浓度为1×104·mL-1,接种于96孔培养板(100 μL/孔),常规培养24 h,Lipofectamine2000与hDOT1L-Scramble、hDOT1L-shRNA质粒制备转染复合物,每孔质粒2.5 μg,转染6 h后,更换新的完全培养基继续培养48 h。按照TUNEL试剂盒说明书进行检测,封片拍照。
1.7 彗星实验检测细胞DNA损伤 收集对数生长期MG63细胞,调整细胞浓度为5×104·mL-1,接种于6孔培养板(2 mL/孔),常规培养24 h,制备Lipofectamine2000与质粒转染复合物,每孔加入质粒2.5 μg,转染6 h后更换新的完全培养基,继续培养至48 h,收集细胞。彗星实验采用三层凝胶结构,第1层为正常熔点琼脂糖,第2层为低熔点琼脂糖和细胞核悬浮液的混合层,第3层为低熔点琼脂糖。裂解时移去每组细胞盖玻片,浸入细胞裂解液,4℃裂解90 min,然后在碱性电泳缓冲液中碱解旋20 min。继续进行单细胞电泳,调整电压为25 V、电流300 mA,电泳10~15 min结束后,每片载玻片上滴加100 μL 30 μg/mL的溴化乙锭溶液,避光染色10 min后在荧光显微镜下观察并拍照,计算拖尾率。利用Comet Assay Software Pect(CASP 1.2.3 beta 1)图像分析软件对彗星图像进行分析。根据拖尾细胞中彗星尾部DNA含量(Tail DNA%),将细胞DNA损伤程度分为5级。0级:无损伤(正常细胞)及细胞损伤率<5%;1级:低度损伤,5%~20%;2级:中度损伤,~40%;3级:高度损伤,~90%;4级:重度损伤,>95%。
1.8 蛋白质印迹分析法检测MG63细胞DOTL1/SIRT1/XPA轴蛋白表达 实验分组及质粒转染方法同前,转染48 h后。收集细胞按照裂解液提取总蛋白后,用BCA蛋白定量试剂盒测定蛋白浓度。取100 μg蛋白加入5×SDS煮沸变性。丙烯酰胺凝胶电泳(PAGE,根据目的蛋白制作10%~15%丙烯酰胺凝胶分离胶和5%浓缩胶),室温封闭2 h,分别加入一抗(DOT1L、SIRT1、XPA、H3K79me2、Histone H3、β-actin,稀释比例均为1∶1 000),4℃孵育过夜,TBST洗膜,加入二抗(1∶2 000)室温孵育2 h,TBST洗涤,滴加ECL显色、定影、扫描免疫印迹。
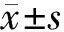
2 结果
2.1 转染后MG63细胞中DOT1L mRNA表达水平 实时荧光定量PCR结果显示,与Scramble组相比,hDOT1L-shRNA组的DOT1L mRNA表达水平明显降低,差异有统计学意义(P<0.01),提示hDOT1L-shRNA转染成功,可用于后续实验。见表1。
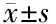
表1 RT-PCR法分析两组MG63细胞中DOT1L mRNA表达量
2.2 敲减DOT1L对MG63细胞的增殖能力的影响 MTT实验结果显示,与Scramble组相比,hDOT1L-shRNA组MG63细胞24、48、72 h时的OD值均降低,差异有统计学意义(P<0.01),见表2。
表2 MTT法检测两组MG63细胞不同时间点的增殖抑制情况
2.3 细胞划痕实验检测敲减DOT1L对MG63细胞迁移能力的影响 划痕实验检测结果显示,与Scramble组相比,hDOT1L-shRNA组MG63细的迁移率降低,差异有统计学意义(P<0.01)。见图1、表3。

图1 DOT1L对MG63细胞迁移能力的影响

表3 细胞划痕实验检测两组MG63细胞迁移率
2.4 TUNEL法检测敲减DOT1L对MG63细胞DNA损伤的影响 TUNEL检测结果显示,与Scramble组相比,hDOT1L-shRNA组MG63细胞的DNA损伤荧光强度明显增高,差异有统计学意义(P<0.01)。见图2、表4。
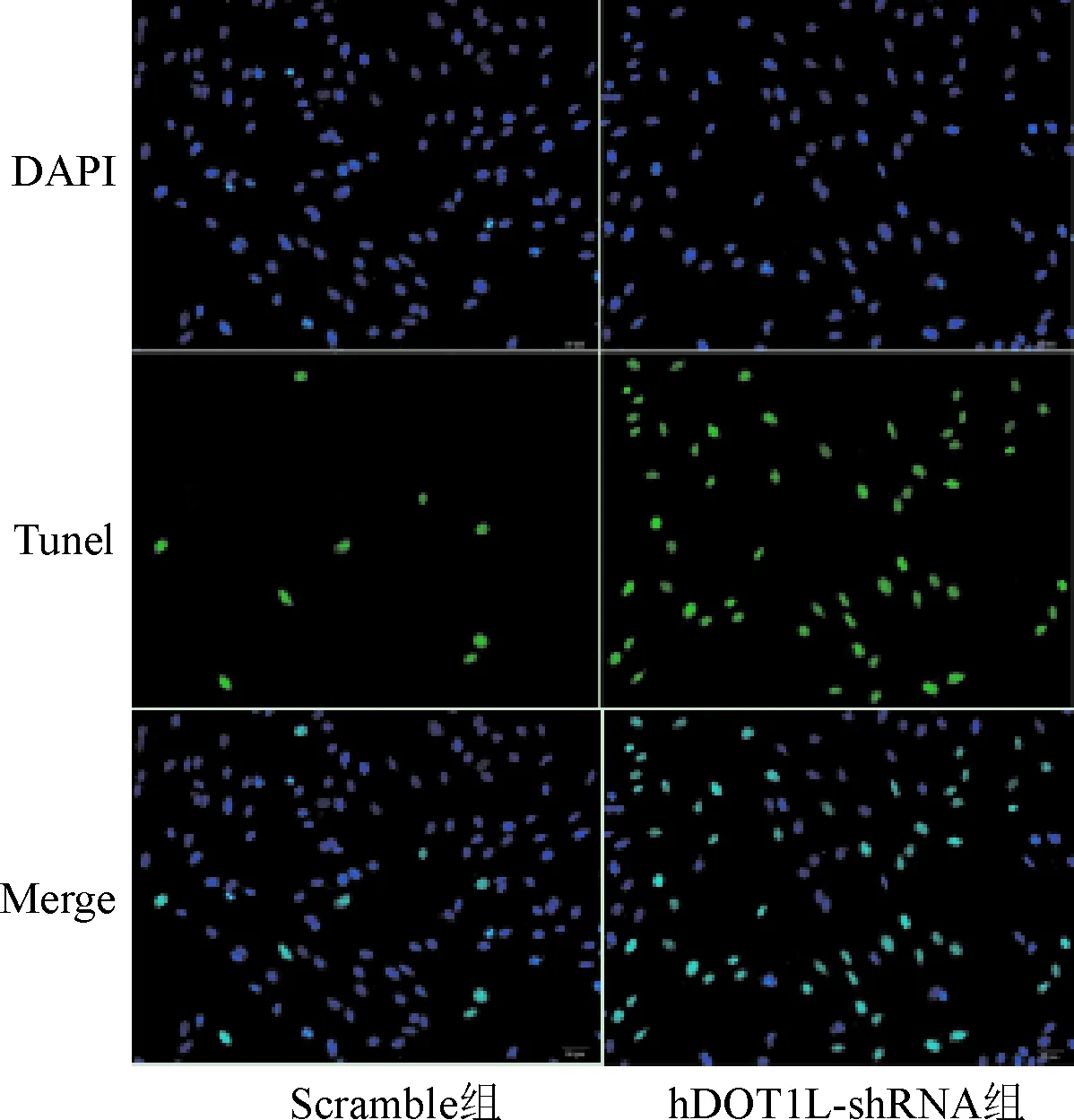
图2 敲减DOT1L对MG63 DNA损伤的影响(TUNEL)
表4 TUNEL法检测两组细胞DNA损伤凋亡情况
2.5 彗星实验检测下调DOT1L对MG63细胞DNA损伤的影响 彗星实验结果显示,与Scramble组相比,hDOT1L-shRNA组MG63细胞DNA损伤率明显上升,差异有统计学意义(P<0.05)。见图3、表5。
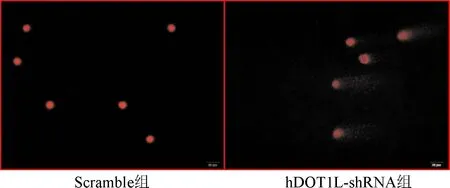
图3 DOT1L对MG63 DNA损伤的影响(彗星实验)
表5 彗星实验检测两组MG63细胞DNA损伤
2.6 DOT1L对MG63细胞DOT1L/H3K79me2-SIRT1L-XPA相关蛋白表达水平的影响 Western blot检测结果显示,与Scramble组相比,hDOT1L-shRNA组MG63细胞中DOT1L、SIRT1、XPA、H3K79me2蛋白表达均降低,组蛋白H3表达总量不变。见图4。
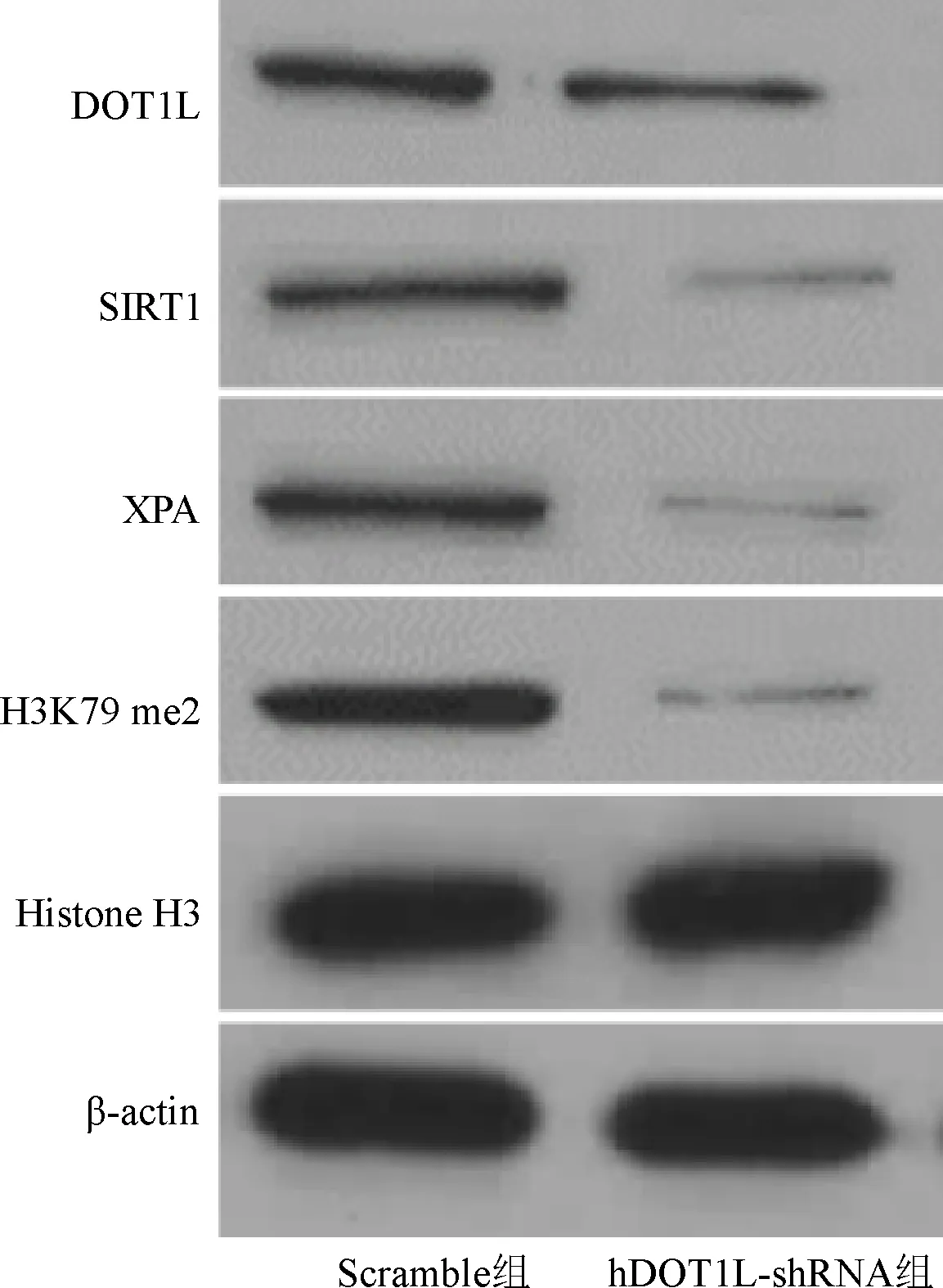
图4 下调DOT1L对MG63 细胞DOT1L/H3K79me2-SIRT1L-XPA相关蛋白表达的影响
3 讨论
骨肉瘤因其高恶性度、高致残率、高复发转移率,耐药性强的等因素导致死亡率居高不下,因此其新药物靶点开发一直是研究热点。最近研究发现组蛋白甲基转移酶在骨肉细胞中异常表达或基因突变,与细胞周期、上皮-间质转分化、细胞凋亡多种机制密切相关,目前发现G9a、EHZ2、NSD家族、PRMT1、PRMT4/CARM1、PRMT6等,主要通过调控组蛋白H3不同位点甲基化调控基因转录,调控骨肉瘤进展[10]。
DOT1L能催化染色质组蛋白H3K79一、二、三甲基化影响转录进程,具有维持细胞基因组稳定的重要功能[6,9]。DOT1L在白血病、前列腺癌、胰腺癌、黑色素瘤等中呈现抑癌/促癌的不同特性[9,11],但骨肉瘤中尚未研究。本研究首次发现敲减DOT1L可抑制MG63细胞增殖和迁移,延迟DNA损伤修复。
SIRT1去乙酰化p53、FoxO3a、Ku70、WRN、NBS1等多个DNA双链损伤相关蛋白,影响其稳定性及活性[12-13]。骨肉瘤中SIRT1基因表达增高,参与骨肉瘤EMT介导的细胞迁移[8],调控SIRT1可改善骨肉瘤的化疗后耐药性[14]。小鼠肾内髓质上皮细胞mIMCD3中SIRT1蛋白与DOT1L相互结合,促进DOT1L对H3K79甲基化[15],表明SIRT1可能参与了DOT1L催化H3K79双甲基化促进DNA修复。
XPA基因是核苷酸切除修复(NER)过程必须,其缺失将导致细胞内NER进程阻滞、突变积累,基因组稳定性下降,最终导致癌症发生或复发,提示XPA可能是NER过程的限速酶之一[16]。此外,XPA基因突变导致XPF和XPG在DN中结合方式改变,由持续变为间断机制,酶活性受到限制,抑制NER途径修复[17]。在UV诱导的DNA损伤修复中,SIRT1通过调控XPA乙酰化/去乙酰化,并与乙酰化XPA形成蛋白复合体[18]。
综合上述分析得到科学假设:DNA损伤后启动修复中,DOT1L、SIRT1、XPA形成级联调节。具体机制是:DOT1L催化H3K79 me2交联与核小体上,继而招募SIRT1蛋白与DOT1L蛋白形成复合体,再招募乙酰化XPA蛋白相互结合,即DOT1L/H3K79me2-SIRT1-XPA级联调节。
本研究通过敲减DOT1L发现H3K79me2、SIRT1、XPA蛋白降低,提示DOT1L可能通过本信号级联调节调控骨肉瘤细胞的增殖、迁移和DNA损伤。但上述蛋白的相互结合,应通过免疫共沉淀研究证实,是本研究未完善之处。此外,本研究中未设空载质粒对照对照组,系因项目组shRNA TR系列质粒购自美国GE公司,含Scramble质粒1个,载shRNA质粒5个,未提供空载质粒,本研究经PCR验证敲除效率选取效率最佳者进行后续研究,对照组仅使用对照更加严格的Scramble乱序对照,保证实验结果可靠性。
综上所述,敲减DOT1L基因表达,可抑制骨肉瘤细胞的增殖、迁移能力,诱导DNA损伤加剧,其机制是通过调控DOT1L/H3K79me2-SIRT1-XPA信号通路实现,但这一作用机制有待进一步通过免疫共沉淀等技术,以及在体内和体外实验证明,这是本研究尚需完善之处。
猜你喜欢
西湖(2022年10期)2022-10-19
小哥白尼(趣味科学)(2022年5期)2022-08-15
江西农业学报(2021年4期)2021-04-20
小聪仔(科普版)(2020年12期)2021-01-18
小学科学(2019年7期)2019-08-27
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
癌变·畸变·突变(2015年3期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
西南军医(2015年6期)2015-01-23
