
荧光碳点的制备、发光机理及应用
2021-09-03 00:28:16汪夏燕孙再成
发光学报 2021年8期
张 震,曲 丹,安 丽,汪夏燕,孙再成
(北京工业大学化学与生物系 绿色催化与分离北京市重点实验室,环境安全与生物效应中心,北京 100124)
1 引 言
2004年,Scrivens课题组的研究人员通过电弧放电法制备单壁碳纳米管时,首次观察到具有明亮荧光的碳纳米粒子[1]。两年后,Sun等首次合成了不同尺寸的稳定发光碳纳米颗粒,并首次将其命名为“碳点(CDs)”[2]。碳点自发现以来就受到了广大科研人员的关注,其优秀的发光性能和良好的生物相容性在生物成像、检测等领域有着重要的应用潜力[3]。碳点是一种尺寸小于10 nm的类球形具有荧光性质的碳纳米颗粒,这种小尺寸的荧光材料发光范围可调,光稳定性能良好,并且对环境友好,低毒性,使其成为传统半导体量子点的理想替代材料。本文主要总结了近年来碳点的制备方法,并对碳点在生物成像[4-6]、检测、化学分析等方面的实际应用进行了综述。
2 碳点的制备方法
自从碳点被发现以来,人们对碳点的认识不断加深,提出了一系列制备优异性能碳点的方法,这些方法可以分为两大类:自上而下法[7-8]和自下而上法[9]。如图1所示。
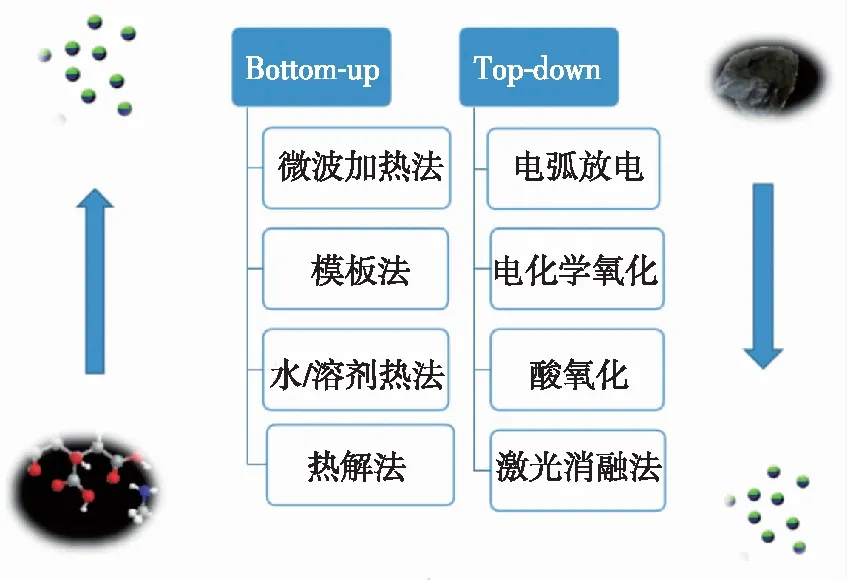
图1 CDs在调控植物不同生理过程中的作用[13]
2.1 自上而下法
自上而下法就是通过对大的碳材料进行剥离或者破碎形成小的碳纳米颗粒,然后通过对碳纳米颗粒的表面进行修饰,提高其发光效率的一种方法,主要包括电弧放电法[1]、激光消融法[10-12]、电化学氧化法[13-16]、酸氧化法[17-20]。
2.2.1 电弧放电法
2004年,美国南卡罗莱纳大学的Xu等[1]报道了利用电弧放电来制备单壁碳纳米管的方法,在电泳法纯化产物的过程中首次发现了可以发出明亮荧光的未知碳纳米颗粒。电弧放电法是最早的制备碳点的方法,他们在电弧放电烟尘中纯化单壁碳纳米管的过程中得到两种新型纳米材料,其中一种成分是短的管状碳,另一种成分是从碳纳米管中提取的荧光纳米颗粒的混合物。对混合物进行分离,得到了可以在365 nm紫外光激发下分别发蓝绿色、黄色和橘色的荧光碳点。两年后,Bottini等[21]将荧光纳米颗粒从原始碳纳米管和硝酸氧化碳纳米管中分离出来,荧光纳米颗粒的荧光发射波长随着纳米颗粒分子量的增加而逐渐红移,得到了荧光发射从蓝色到黄绿色的碳点。电弧放电法制备碳点的过程中,由于电弧放电灰的成分复杂,杂质较多,因此制备的碳点难以分离纯化,产率很低,荧光量子效率也很低;但是优点是制备出的碳点粒径较小,含氧量较高。
2.1.2 激光消融法
2006年,Sun课题组[2]通过在氩气氛中用Nd∶YAG激光器在900 ℃和75 kPa的条件下烧蚀碳靶得到了碳纳米颗粒,样品在水悬浮液中并没有观察到荧光,作者将其在硝酸水溶液中处理后仍没有观察到荧光。然而,通过将简单的有机物种附着在酸处理的碳颗粒表面钝化,得到了可以产生明亮荧光的碳纳米颗粒,并首次将其命名为碳点。通过研究发现制备出的碳点的发光波长会随着激发波长的变化而变化,展现出从蓝到红的荧光发射。此后,Yang等[22]通过相同的方法,以13C作为碳源,制备得到了平均直径4~5 nm的碳点,并且通过不断优化实验参数使量子产率达到20%。2009年,Hu等[23]以碳粉悬浮液为原料,在有机溶剂中通过激光照射合成了荧光碳纳米颗粒。这种方法可以在形成聚合物的同时对碳点进行表面改性,作者发现选择不同的溶剂可以调节碳点的荧光发射波长。2011年,Li等[24]提出了一种制备碳量子点的简单方法,如图2所示,即在有机溶剂中对碳纳米粒子进行激光快速钝化。制备的碳点具有明亮的、可调谐的、稳定的光致发光特性。使用激光消融法制备碳点通常需要有照射、氧化及钝化过程,制备方法复杂,碳点产量低,粒径分布不均匀,纯度较低。

图2 简化后的激光消融法制备碳点示意图[24]
2.1.3 电化学氧化法
电化学氧化法是通过电解一些碳材料,如碳纳米管、石墨等来制备碳量子点的方法。2007年,Zhou等[13]以0.1 mol·L-1四丁基高氯酸铵(TBAP)-乙腈溶液为支撑电解质,首次利用多壁碳纳米管作为工作电极,以铂网和Ag/AgClO4分别为对电极和参比电极,通过循环伏安法处理多壁碳纳米管工作电极,得到了平均粒径大约为2.8 nm的碳点。由于多壁碳纳米管是由滚动的石墨烯层(SI)形成的,作者认为在电化学循环过程中,四丁基高氯酸铵阳离子很可能插入到多壁碳纳米管缺陷中,破坏并剥离缺陷附近的碳纳米管,进入电解质溶液中形成碳点。Zhao等[14]在2008年通过一种简单的电化学氧化法制备出了碳点,他们以石墨作为工作电极、饱和甘汞电极作为参比电极、铂线作为对电极,在0.1 mol·L-1的NaH2PO4水溶液中进行电氧化,制备出了尺寸为1.9 nm和3.2 nm的荧光碳点。经过研究发现制备出的碳点随着直径的增加,荧光发射峰从445 nm红移到510 nm。通过高分辨透射电镜(HRTEM)表征发现碳点的晶格间距为0.328 nm,具有与石墨类似的结构特征。此后,Zheng[15]、Lu[16]等均通过电化学氧化的方法制得了荧光碳点。2010年,Li等[25]以石墨棒为正极和负极、NaOH/EtOH为电解液,报道了一种简单的一步碱辅助电化学制备的具有尺寸依赖光致发光(PL)和优异上转换发光性能的碳点,碳点的尺寸在1.2~3.8 nm之间。作者通过以酸性溶液(如H2SO4/EtOH)为电解液的一系列对照实验发现碱性环境在生成碳点的过程中起到了十分关键的作用,大量的OH-能够促进碳点的生成,并且通过改变电流的密度,制备出了不同发射波长的碳点。值得一提的是,Ming等[26]提出了一种简单的电化学方法,用于大规模合成高纯度高质量的碳点,实验装置如图3所示。该方法只使用纯水作为电解液,不使用任何其他化学添加剂,所制备的碳点具有高结晶性、良好的水分散性、显著的光致发光性能,并且无需进一步纯化,为大规模制备碳点提供了一种新的途径。电化学氧化法制备碳点的优点是可以通过改变电流强度从而调节碳点的尺寸及其发光性质,并且制备成本低,产率高;缺点是产物的荧光量子效率较低。
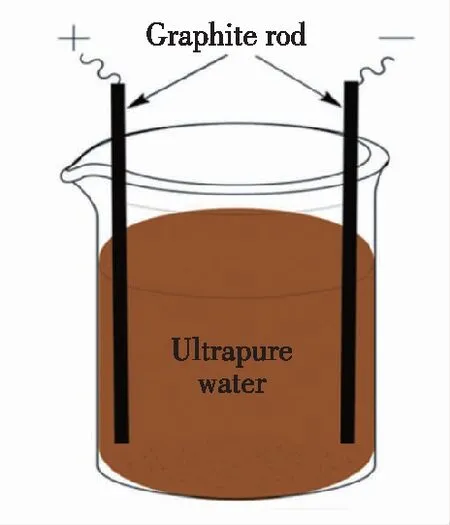
图3 电化学氧化法制备碳点示意图[26]
2.1.4 酸氧化法
酸氧化法主要是通过酸处理碳源[8],使碳源表面的官能团氧化从而可以产生荧光。2007年,Liu等[27]通过酸氧化蜡烛燃烧产生的烟灰制备了多色荧光碳点,用聚丙烯酰胺凝胶电泳法对制备的碳点进行了纯化。作者认为酸处理有以下几个作用:(1)将碳聚集物分解成小的纳米颗粒;(2)增加碳纳米颗粒的溶解性;(3)影响碳点的荧光性质。2009年,Ray等[28]通过相同的方法制备出碳点,他们将蜡烛烟灰与5 mol·L-1的硝酸在回流的条件下反应12 h,得到尺寸2~6 nm的碳点。并且他们发现蜡烛烟灰与硝酸回流在12 h内碳点的荧光量子产率随时间延长而增加,回流超过12 h则对碳点的荧光量子产率没有影响。文献[29]将天然气燃烧烟尘在硝酸中回流制备得到碳点,作者通过核磁共振和红外光谱对制备的碳点进行了表征,证实了以芳基和羧基/羰基的形式存在sp2碳,这表明光致发光可能来自粒子的表面态。Guo等[30]通过硝酸氧化活性炭得到了平均直径为5 nm的具有绿色荧光的碳点,同时用硼氢化钠后处理还原碳点发现其荧光量子产率从2%提高到了11%。作者分析化学还原增加了碳点上羟基的数量,同时减少了羰基的数量,从而增加了表面态发光,提高了荧光量子产率。酸氧化法制备碳点方法简单,实验设备要求较低,但是制备碳点的产率普遍偏低,分离较为困难,并且一般需要对碳点进行后处理钝化。
2.2 自下而上法
2.2.1 热解法
热解法制备碳点主要是以有机物作为碳源,在高温下热解碳化从而形成碳点。2008年,Giannelis团队[31]通过高温热解柠檬酸铵盐一步得到了碳点,通过后续的研究发现碳点的荧光特性与其表面与酰胺相关的官能团有关,并且过高的反应温度会导致碳点的聚合,从而降低碳点的产率与荧光量子效率。2010年,Wang等[32]通过在氩气气氛下将柠檬酸在LiNO3熔融盐中热氧化反应制备出了白光碳点,其荧光量子产率为53%。由于碱金属硝酸盐的熔点较低,与大多数固态反应相比,熔融硝酸盐中的反应是软化学的一个分支,这也是在温和条件下制备碳点的一个先例。此后,Wang等[33]首次用有机硅烷作为配位溶剂和功能化试剂,通过热解有机硅烷和有机酸的混合物在短时间内一步制备了硅烷功能化的无定形碳点,制备过程如图4所示。这种碳点具有良好的生物相容性,荧光量子产率可达47%,该合成路线为碳点的制备提供了一种新的思路。除了以一些有机酸作为反应的原材料,Liu等[34]在浓硫酸条件下,140 ℃热解乙二醇一步碳化合成了水溶性石墨化碳点。在没有钝化的情况下,其荧光量子产率达到了25%,将反应温度降低至80 ℃,其荧光量子产率大幅度提高至62.9%,说明温度对热解制备碳点起到了一定的调节作用。该方法制备的石墨化碳点对水中的Hg2+具有良好的选择性和敏感性。热解法制备碳点碳源种类广泛,例如己烷[35]、柠檬酸和乙二胺[36]、马来酸酐和四乙烯五胺[37]等,这种方法制备过程简单,可以通过调整热解温度调节碳点的发光性质;并且相对于前面所提到的制备方法,热解法制备碳点的荧光量子效率较高。
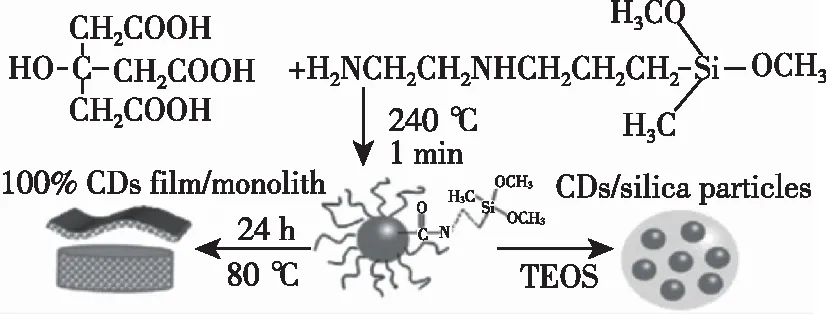
图4 光致发光CDs、柔性CDs薄膜和CDs/二氧化硅颗粒制备原理图[33]。
2.2.2 水/溶剂热法
水热法是通过将碳源与溶剂混合、在高压釜中加热制备碳点的一种方法,值得一提的是,水热法是目前最常用的制备碳点的方法。2010年,Zhang等[38]使用L-抗坏血酸为碳源,采用水热法直接制备了荧光碳点,合成的碳点具有较高的光致发光效率(6.79%),其直径约为2.0 nm。作者发现通过水热法制备的碳点既不需要用强酸处理,也不需要表面改性即可产生荧光,并且在水溶液中至少可以稳定存在半年以上。此后,Yang等[39]在磷酸二氢钾存在下,利用葡萄糖水热处理合成了荧光碳点。通过简单地调整磷酸二氢钾的浓度,制备了荧光发射可调谐的碳点。作者将制备的碳点应用于细胞毒性测试,在碳点浓度达到0.625 mg·mL-1时,细胞存活率在72 h后仍高达95%,说明碳点具有较小的生物毒性。2012年,Chang课题组[40]经过研究发现含氨基和羧酸基团的化合物适合制备高光致发光和水溶性的碳点。他们通过实验提出了碳点的生成要经历4个阶段:脱水、聚合、碳化和钝化,如图5所示。最初,反应物分子由于氢键作用力产生聚集;然后,在加热(脱水)过程中,聚合发生,生成小的碳核;随着反应时间的增加反应物分子向碳核表面扩散,产生的碳核随之增大,最终生成碳点。Lu课题组[41]开发了一种简单、方便、可控的单反应体系水热制备CDs的方法。通过控制反应温度等条件,合成了具有尺寸效应的碳点,随着尺寸的增大,激发波长会逐渐红移。许多天然的生物质比如橙汁[42]、柚子皮[43]、烟丝[44]、红豆杉[45]等都可以被用来制备碳点。水热法制备碳点,原材料来源十分广泛,反应设备简单,反应条件易控,并且由于反应在密闭的反应釜中进行而避免了空气中杂质的影响,制备的碳点较为均匀。水热法合成的大部分碳点都具有良好的水溶性,但是由于水热釜容积有限,一次性制备碳点的量有限,并且水热法制备碳点会产生较多的杂质,导致分离提纯碳点困难,不适合大规模工业化生产。
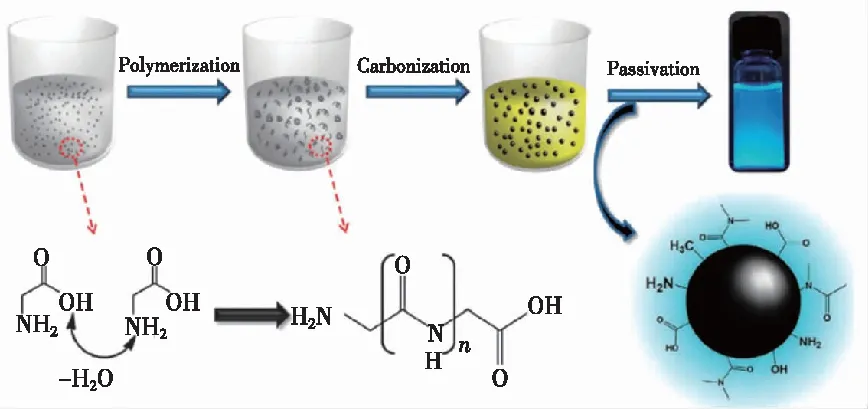
图5 甘氨酸通过脱水、聚合、碳化和钝化合成碳点示意图[40]。
2.2.3 微波加热法
Yang课题组[46]于2009年采用一种新的方法合成了发射绿色荧光的碳点。他们将聚乙二醇与糖类在水溶液中混合,然后通过微波辅助加热合成了量子产率6.3%~3.1%的具有激发依赖行为的碳点。微波加热法合成碳点是利用微波在短时间内快速升温使反应物单体聚合碳化从而生成碳点的一种方法,为碳点的合成提供了一种新的快速的方法[47]。Peng等[48]用糖类作为反应的碳源,经4,7,10-三氧-1,13-十三烷二胺(TTDDA)、乙二胺、油酰胺或PEG1500在氮气氛围下钝化72 h后也获得了荧光碳点。体系中不添加钝化剂也可以生成碳点,但是生成的碳点不规则,且荧光强度较弱。Wang等[49]在此基础上对微波加热法进行了改进,以碳水化合物(甘油、乙二醇、葡萄糖、蔗糖等)和少量无机离子为原料,无需钝化剂,在几分钟内即可得到碳点。同时,作者发现加入三价离子会提高碳点的产率及荧光量子效率。Jiang等[50]将组氨酸溶解在磷酸溶液中通过微波加热法制备出荧光量子产率高达44.9%的蓝色荧光碳点。Sun等[51]将柠檬酸与甲酰胺溶液混合后微波加热,制备了发射波长约为640 nm的碳点。微波加热法制备碳点具有方便快捷、制备条件简易、原料易得等优点,但是仍有反应过程不稳定、反应温度不易控制、荧光量子产率较低等缺点。
2.2.4 模板法
碳点作为一种纳米尺寸的碳化量子点,在合成的过程中十分容易团聚,而模板法则可以有效地避免碳点团聚,形成尺寸均匀的碳点。2009年,Liu等[52]首次提出了通过模板法制备碳点。首先,他们利用两亲性三嵌段共聚物F127修饰硅胶球体作为载体,以可溶性酚醛树脂作为碳源,制备出球形聚合物/F127/二氧化硅复合物;随后通过高温处理并用氢氧化钠去除二氧化硅载体,再经过酸处理和表面钝化,制备过程如图6所示,制备出了尺寸在1.5~2.5 nm的碳点,荧光量子产率为14.7%。Zong等[53]以介孔二氧化硅球为纳米反应器,采用浸渍法制备亲水性碳点,他们将二氧化硅球浸入到柠檬酸和盐的溶液中,经过高温煅烧、氢氧化钠刻蚀、透析得到了尺寸在1.5~2.5 nm的荧光碳点。2017年,Gu等[54]通过将葡萄糖分子装入金属有机骨架(MOFs)的孔隙、加热到200 ℃生成了碳点。CDs的大小和间距可以通过不同模板MOF来控制,比如HKUST-1、ZIF8或MIL-101,得到的CDs尺寸约为1.5,2.0,3.2 nm,与相应的MOF孔径尺寸相近。该方法合成的碳点具有良好的发光性能,发射波长随着尺寸的增加而红移。模板法合成碳点步骤如下:首先在模板上合成碳点,然后通过加热、酸(或碱)刻蚀等手段除去模板。模板法合成碳点的优点在于可以通过模板的大小控制碳点的尺寸,并在合成的过程中减少碳点的聚集,制备的碳点尺寸均匀,荧光量子产率较高;缺点是有些模板与碳点难以分离,并且在酸碱刻蚀或者加热去除模板的过程中有可能会影响碳点的荧光性能。
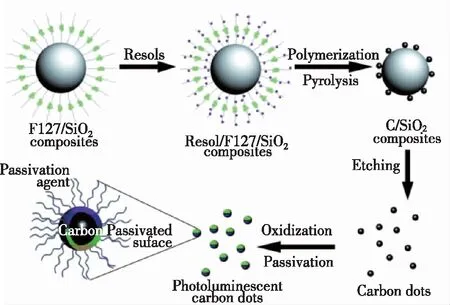
图6 多色光致发光碳点合成示意图[52]
2.2.5 其他方法
随着对碳点的研究不断深入,除了以上合成方法,近年来发展出一些新的碳点制备方法。 2011年,Li[55]以活性炭为原料,与过氧化氢混合后采用超声法一步合成了具有优良的上转换荧光性能的水溶性荧光碳点。2016年,Chen等[56]以对苯二酚和乙二胺为前体,通过乙二胺催化过氧化氢在室温下分解放出大量热,从而合成了荧光量子产率为24.6%的碳点。后来,Chen等[57]将对苯二酚与过氧化氢在室温下混合,再与三乙基四胺反应,通过反应过程中自发放出的热量合成出了平均尺寸为4.5 nm的碳点。自放热反应合成碳点具有反应原料易得、反应时间短、装置简单的优点,并且合成过程没有过多的能量损耗,节约能源,碳点产率较高,近年来受到了越来越多的关注。
3 碳点的发光机理
实验中,由于使用的碳源不同,因此合成的碳点光学性质差别也较大,不过通常条件下碳点在270~360 nm的紫外光区有一个或者多个吸收峰。碳点与量子点类似,带隙通常在可见光范围内,因而碳点具有光致发光现象,这也是碳点最具有优势的特征。但是,因为碳点的原料来源广泛,制备方法多,其结构较为复杂,发光机理多种多样,并不能完全解释清楚[58]。目前,关于碳点的发光原理分为内部主导因素与外部主导因素。内部主导因素主要有以下三种解释:(1)由Bourlinos等[31]提出,他们通过热分解有机物得到碳点,认为碳点的发光是由于形成了多种芳香化合物所导致的。有机化合物在热解的过程中出现了碳原子的重新组合,形成与石墨氧化物等类似的多环芳香化合物,引起碳点发光。(2)由Zhao等[14]提出,他们通过一种简单的电化学氧化法制备出了尺寸不同的碳点,发现随着碳点直径的增加,荧光发射峰逐渐红移,因此认为碳点的荧光发射与量子点相似,是由尺寸决定的,但是有许多情况会违反这种依赖性。2018年,本课题组[59]以柠檬酸和尿素作为反应原料,改变反应温度以及反应物的配比,通过水热法制备了从蓝色一直到红色发射的系列荧光碳点,如图7所示。通过研究发现碳点发射波长的变化与碳点石墨化程度和表面官能团的含量有关,发射波长的红移是石墨化程度的增加或有效共轭长度的增大所致,而不是由于尺寸的增大,这一研究为制备不同发射波长的碳点提供了指导。(3)Sun等[60]认为碳点的发光是由于碳点在合成的过程中产生了表面的能量陷阱,通过表面修饰后即可产生荧光发射,主要考虑两类表面状态,即表面构型和掺杂原子[61]。除了以上三种解释之外,协同作用促使碳点发光更容易被接受[62],即碳点的发光是由于多种作用相互协同所导致的。一些研究显示碳核与碳点的表面态都是碳点发光的原因[63-64],也有研究表明碳点的共轭作用与碳点表面的缺陷是导致碳点发光的原因[65]。而外部主导因素主要是由于分子态和环境效应所引起的[56],碳点合成过程中的分子态是影响其发射的一个重要因素,而环境所导致的温度、压力、PH值等差异同样会影响碳点的发光。目前对碳点的研究逐渐深入,但是由于合成碳点的碳源种类繁多,影响碳点合成的因素过多,合成的碳点发光性质各不相同,因此对碳点发光机理的解释还并不完善,尚需更深入的研究和更核心的理论提出。
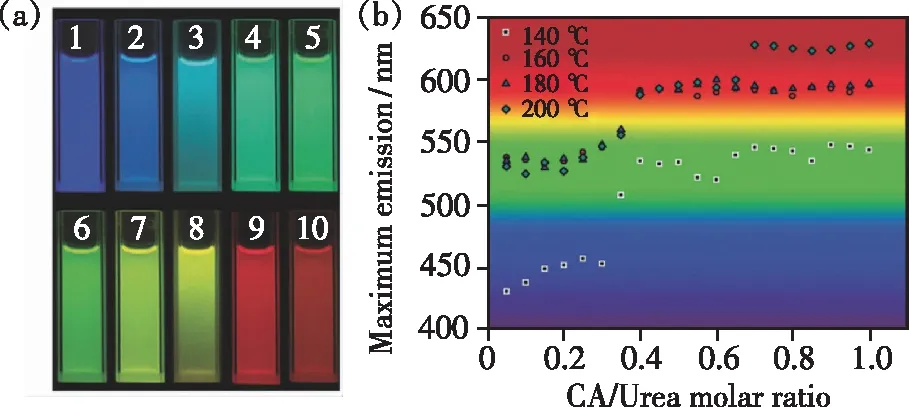
图7 以柠檬酸和尿素为原料制备的碳点乙醇溶液在不同反应条件下的光学性质。(a)不同反应条件下制备的发光碳点在不同激发光下的光学图像;(b)不同柠檬酸/尿素量比和不同反应温度下碳点的最大发射峰[59]。
4 碳点与碳点复合材料的应用
碳点相对于传统的半导体发光材料具有更加优良的应用潜力,由于碳点在合成的过程中没有重金属参与反应,因此碳点材料具有无毒、生物相容性良好、对环境污染小的优点。碳点在合成的过程中通过控制反应条件,可以调控其尺寸大小和发射波长,具有重要的实际应用价值。
4.1 生物医学成像
Sun等[2]最先报道了荧光碳点在生物成像中的应用。他们用PEG1500钝化碳点,然后与一定浓度的大肠杆菌悬液混合;在无菌的试管中,室温下旋转18 h,10 000 r/min离心,得到用碳点标记的细胞悬液,作者用不同波长激发光照射得到了具有不同发射的细胞图像。Xu等[66]以l-胱氨酸和邻苯二胺作为原料通过水热法制备出了最大发射波长为648 nm的氮硫共掺杂荧光碳点。他们以293T细胞作为模型体系,与碳点溶液混合培养,随着碳点溶液浓度的不断提高,其荧光成像的强度也不断提高,并且当碳点溶液浓度较高时,细胞的存活率仍超过80%,说明荧光碳点不但具有良好的成像能力,其生物毒性也较低,对细胞的伤害较小。Zuo等[67]将4,5-二氟-1,2-苯二胺与酒石酸通过水热反应得到了F掺杂的荧光碳点(F-CDs),其荧光最大发射可达600 nm左右,具有良好的水溶性,并且对广泛的pH变化有很好的稳定性。作者选择了HEK293细胞和B16F10细胞分别作为正常细胞和癌细胞,研究F-CDs的生物成像能力,通过改变F-CDs的浓度得到了清晰的细胞图像,并且当F-CDs的浓度较高时,细胞仍有90%以上的存活率,证明F-CDs具有良好的生物相容性。Su等[68]通过对苯二胺和天冬氨酸的一锅水热处理制备了黄色发射的CDs,其具有优异的光学特性、良好的生物相容性和低细胞毒性,将细胞用碳点处理,得到了清晰的细胞图像。长波长发射的碳点可以很好地避免生物体自荧光对成像清晰度的影响,并且对细胞的影响较小。Yang课题组[45]通过对红豆杉提取物进行处理得到了窄全宽半峰和高深红/近红外发射的CDs。Shan课题组[69]首次采用无溶剂原位碳化法制备了具有57%光致发光量子产率的红/近红外发射碳点,具有良好的生物相容性和生物成像能力。Qu课题组[70]利用富含亚砜/羰基的分子或聚合物对碳点表面进行修饰,得到了红/近红外发射碳点。随着研究的不断进步,红/近红外发射碳点不断被报道,极大地促进了碳点在生物成像领域的发展。
4.2 分子与离子检测
4.2.1 阳离子检测
一般情况下,以阳离子的形式存在的污染物主要是各种重金属离子,如Hg2+、Cu2+、Pb2+、Cd2+、Fe3+[71]和Zn2+等,这些离子存在于环境中,对生物体的健康产生了巨大的威胁,如何检测并特定地识别这些离子是目前比较困难的一个问题。由于碳点容易被电子受体高效猝灭,因此碳点也可被用做阳离子的检测试剂。另外,碳点表面能够被特定的基团修饰(如羧基、氨基和羟基),提高了其对靶点的选择性,可以作为检测特定离子的检测试剂[72],碳点已被越来越多地应用到各种重金属离子的检测中。Cui等[73]开发了基于碳点(CQDs)标记的寡脱氧核糖核苷酸(ODN)和氧化石墨烯(GO)的荧光光谱分析系统,用于水体中汞(Ⅱ)的检测。ODN可以修饰到CQDs表面,形成ODN-CQDs,ODN-CQDs在氧化石墨烯存在的条件下会吸附到氧化石墨烯的表面产生能量转移,导致荧光猝灭。而Hg2+可以促进氧化石墨烯表面ODN-CQDs的释放,从而诱导荧光的恢复,过程如图8所示,通过这一特性可以对Hg2+进行较为精确的检测。Liu等[74]在罗丹明B掺杂二氧化硅纳米颗粒表面包覆CDs,制备了基于CDs的铜离子纳米传感器。他们以N-(β-氨乙基)-γ-氨丙基甲基二甲氧基硅烷(AEAPMS)为主要原料合成荧光CDs,使CDs表面残留的乙二胺基团和甲氧基硅烷基团分别作为Cu2+的识别位点和硅化反应的位点。这种纳米传感器在光激发下显示了罗丹明B(红色)和CDs(蓝色)的特征荧光发射,在与Cu2+结合后,CDs的荧光被猝灭,只剩下罗丹明B的特征荧光,从而检测Cu2+的存在。Song等[75]以氨基水杨酸作为碳源制备了氮掺杂荧光碳点,氮的掺杂不仅极大地提高了CDs的光致发光(PL)效率和发光可调性,而且丰富了碳点表面官能团,可以应用于Fe3+的检测。Wu等[76]制备了可以检测Pb2+的碳点,这种碳点可以与Pb2+形成复合物从而导致碳点的荧光猝灭,以此检测Pb2+的存在。Niu等[77]建立了一种高效的碳量子点/金纳米团簇(CQDs/AuNCs)纳米杂交比值荧光探针,用于对镉(Ⅱ)离子的敏感和选择性检测。此外,Zhang等[78]用喹啉衍生物对碳点表面进行了修饰,使碳点对Zn2+具有高度特异性识别能力,检出限可低至6.4 nmol·L-1。同时,这种Zn2+检测器具有良好的水溶性、生物相容性和细胞膜通透性,可以被用作细胞的成像试剂。碳点表面含有丰富的官能团,这些官能团可以与一些特定的金属阳离子反应,改变碳点的荧光性质,通过碳点荧光性能的变化较为直观地检测金属离子的存在。

图8 基于石墨的Hg2+检测器机理示意图[73]
4.2.2 阴离子检测
4.2.3 分子检测
碳点表面含有丰富的官能团,可以与一些化学物质发生反应,改变碳点的荧光性质,因此碳点可以作为荧光探针来检测特定的化学物质或者小分子,比如氨基酸、四环素[82]等。Shan课题组制备了一种可以检测TCPO-H2O2体系中痕量H2O2和葡萄糖[83]的碳点材料,溶液中的碳点会与葡萄糖和H2O2反应,产生化学发光,具有很高的选择性和灵敏度,检出限分别为11.7 μmol·L-1和12.6 μmol·L-1。碳点还可以用于检测氨基酸,Luo课题组[84]利用槲皮素和乙二胺制备了一种对pH敏感的荧光碳点,研究发现pH值的变化会造成碳点聚集导致荧光猝灭,作者将其应用于天冬氨酸和谷氨酸的检测。碳点也可以用于DNA的检测,Gomez课题组[85]利用海藻作为前驱体,采用水热法制备了氮掺杂碳点,合成的碳点在凝胶电泳中可以作为优良的荧光载体检测DNA条带,是一种良好的核酸标记试剂。碳点与传统的检测试剂相比具有良好的选择性与灵敏度,对生物体的毒性更小,对环境的污染小,具有很大的应用潜力,但是限制其应用的仍然是无法大规模工业化生产。
4.3 催化应用
4.3.1 光催化
光催化氧化还原技术是一种新型、绿色的化学催化技术,在新能源等方面具有重要的应用前景。碳点具有光诱导电子转移[86-87]、氧化还原、可见光吸收[88-90]等优良性质,并且毒性低,对环境友好,同样具有良好的发展前景。2010年,Kang等[25]报道了一种简单的一步碱辅助电化学制备的具有尺寸依赖光致发光(PL)和优异上转换发光性能的CQDs,其尺寸为1.2~3.8 nm。他们用溶胶-凝胶法制备了二氧化钛/碳点(TiO2/CQDs)和二氧化硅/碳点(SiO2/CQDs)复合材料,并在光照下有效降解了亚甲基蓝。他们进一步解释了光催化反应的过程。当二氧化钛/碳点或二氧化硅/碳点纳米复合光催化剂被光照射,碳点吸收长波长的光,然后发出波长较短的光作为上转换的结果,进而激发二氧化钛或者二氧化硅形成电子空穴对。然后,电子转移到碳点上,空穴留在二氧化钛或者二氧化硅上,产生有效的电子空穴对分离,从而降解亚甲基蓝,机理如图9所示。随后,该课题组将碳点嵌入到氮化碳(C3N4)中[91],制备出了一种无金属碳纳米点-氮化碳(C3N4)纳米复合材料,这种复合材料在光催化太阳能水分解方面具有优异的性能。在入射光波长为λ=(420±20) nm时,量子效率为16%;入射光波长为λ=(580±15) nm时的量子效率为6.29%;入射光波长为λ=(600±10) nm时的量子效率为4.42%。碳点的加入拓宽了C3N4对可见光的吸收范围,并且碳点-氮化碳复合材料具有很高的稳定性,在超过200天的200次循环使用后仍保持稳定性。我们课题组[92]以柠檬酸和尿素粉末为原料,在控制温度下,通过一步均相热裂解将碳点结合到氮化碳中,合成了g-C3N4-C光催化剂。通过控制反应温度,对反应的过程进行了分析,发现在200~300 ℃的温度范围内,带有封端尿素的CDs可以在熔融的柠檬酸/尿素溶液中形成。因此,CDs在最终的g-C3N4-C复合材料中的负载可以容易地通过柠檬酸的添加量来控制。合成的g-C3N4-Cx复合材料光催化剂的带隙通过控制柠檬酸的负载有效地从2.84 eV微调到2.08 eV。通过追踪光生电子和空穴的转移路线,我们发现光生电子被转移到CDs中,相应的空穴保留在g-C3N4上,证明了CDs在g-C3N4-Cx复合物中具有两种功能。一个功能是产生H2的电子受体和助催化剂,另一个功能是分解水氧化产生的H2O2的催化剂。这项工作为设计更稳定和高效的无金属光催化剂提供了思路。Tang课题组[93]通过微波法制备了一种具有独特的空穴接受特性的碳点,添加到氮化碳中,可以延长氮化碳的电子寿命,更有可能被用于吸附质子和电子转移[94],这种催化剂在光照下可以在水溶液中将二氧化碳转化成醇类。这项工作也证明碳点在光催化应用中的独特作用。

图9 TiO2/CQDs在可见光下的催化机理[25]
4.3.2 电催化
碳点具有高比表面积、优良的导电性、高孔隙率以及吸附能力,使其在电化学方面成为潜在的优良材料。Yang等[95]通过一步电沉积金属硝酸盐和氮掺杂碳点(N-CQDs),制备了亚纳米NiFeOx团簇和氮掺杂碳点的高耦合杂化材料。由于NiFeOx团簇与氮掺杂碳点之间存在较大且高度耦合的界面,NiFeOxNC杂化材料表现出优越的电催化析氧性能(OER),作者将其与商用的RuO2催化剂和DSA电极进行了比较,证明其具有较低的过电位和更快的动力学,以及在实际操作条件下良好的长期耐用性和环境适应性。Xiong课题组[96]提出了一种简单的利用CDs制备Co9S8/碳杂化材料的策略,通过将CDs和Co9S8纳米粒子包埋在N和S共掺杂的碳基体中,获得了Co9S8/CD@NSC材料,制备过程如图10所示。CDs的引入降低了Co9S8纳米粒子在合成过程中的生长和聚集,促进了其在碳基体中的分布,最终提高了产物的比表面积和电导率。Co9S8/CD@NSC对氧还原反应(ORR)和析氧反应(OER)均表现出优异的催化活性和长期耐用性,其催化性能优于单纯的Co9S8@NSC、商业Pt/C电极和RuO2催化剂。这项工作为非贵金属基ORR和OER双功能催化剂的合成提供了新的思路。过渡金属磷化物(TMPs)具有催化效率高、成本低等优点,是催化析氢反应(HER)的良好电催化剂。Chen等[97]在TMPs表面沉积碳量子点(CQDs),通过增加TMPs表面积和活性位点数量提高了催化析氢的性能。此外,其在酸和碱溶液中也能稳定存在。

图10 Co9S8/CD@NSC和Co9S8@NSC的制备程序示意图[96]
4.4 碳点复合材料的应用
目前有很多碳点复合材料的报道,值得一提的是碳点的磷光材料。生物体中由于含有种类繁多的化学物质,比如核黄素、叶酸、酪氨酸、色氨酸等,在紫外光照射时会产生很强的自体荧光现象[98],因此,可以通过提高成像剂的发射波长或者通过延时发光来避免自体荧光的影响,提高成像的精确性。2013年,Song等[99]将四乙酸二钠盐在高温下反应得到的碳点与聚乙烯醇(PVA)溶液混合干燥后首次观察到了磷光现象,作者分析是碳点表面的官能团与聚乙烯醇形成了氢键,通过氢键的固定作用在一定程度上限制了碳点表面官能团的振动与转动,稳定了三重激发态,从而产生磷光的现象。一般与碳点复合产生磷光的基体有沸石[100]、硅胶、聚合物等,碳点的磷光材料与碳点一样具有发光波长和尺寸可调的优点,并且由于磷光发光时间长、信噪比高等特点,因此在生物成像领域具有很大的应用潜力,成为目前研究的热点。
Liu等[101]通过将碳点与二氧化硅负载,制备得到了磷光寿命1.64 s的磷光材料CDs@SiO2,在紫外光激发下,其延时发光肉眼可见可达10 s。作者分析是碳点表面的官能团与硅胶生成的C—O—Si共价键稳定了三重激发态,从而产生长寿命的磷光。他们还将磷光材料与豆芽共同培养,从而将磷光材料引入到植物体中,在紫外灯关闭之后,经过处理的豆芽仍能观察到明亮的磷光,如图11所示,从而避免了生物体自体荧光对成像的影响。Shan等[102]将碳点(CNDs)采用二氧化硅封装,制备了二氧化硅碳点材料(WSP-CNDs@silica),二氧化硅的包覆既保证了CNDs的溶解性,还将CNDs与水溶液中溶解的氧隔离开来,碳点与二氧化硅生成的共价键稳定了三重激发态,促进了长寿命磷光的产生。作者对WSP-CNDs@silica进行了细胞毒性测试,当WSP-CNDs@silica的浓度增加到100 μg/mL,培养48 h后,细胞的存活率仍在90%以上。同时作者用WSP-CNDs@silica对DC2.4细胞进行了生物成像测试,发现碳点材料可以很好地被细胞吸收,并能够在光源关闭后产生很强的磷光,获得了细胞清晰的成像。作者进一步使用WSP-CNDs@silica进行了小鼠活体成像测试,体内余辉成像采用IVIS活体成像系统,将碳点材料注射到小鼠背部的皮肤下,通过紫外光源激发,收集到了很明显的磷光信号,这说明WSP-CNDs@silica材料可以作为生物成像试剂。Li等[103]将丙烯酰胺加入到柠檬酸尿素体系中,制备出了具有良好水溶性的碳点,经过研究发现其具有磷光性质,作者将其添加到墨水中制备出了新型防伪墨水,可应用于信息加解密、高级防伪和复杂图案的隐藏。近年来对磷光碳点材料的研究不断发展,极大地拓展了碳点在生物成像[104]、信息加密[105]、光电器件[106]等领域的应用潜力,但目前的磷光碳点材料很少有长波长磷光发射的报道,如何增强其发射波长,使其具有更强的穿透能力仍是其面临的主要挑战之一。并且磷光碳点材料的延迟发光时间要比其荧光长很多,如何进一步增加延迟发光的时间也是科研人员追求的目标之一。

图11 未经处理的豆芽(左)和经过CDs@SiO2处理的豆芽(右)在365 nm紫外光照射中和关闭紫外光之后的对比图[101]
除了磷光碳点材料,还有许多其他的碳点复合材料,比如碳点在电极材料中的应用[107]。硬碳材料具有较好的性价比,被认为是最有前途的钠离子电极材料。然而,与锂离子电池石墨电极相比,其相对较低的比容量和初始库伦效率(ICE)仍然限制了其进一步发展。Xie等[108]利用水热碳化的方法,将制备得到的碳点制作成电极并应用到钠离子电池的阳极上,其初始库伦效率为91%,是目前报道的最高值之一。碳点由于其出色的光致发光性能,在LED[109-112]、指纹检测[113]等方面同样具有巨大的应用潜力。一些碳点复合材料具有良好的生物相容性与低毒性等优点[114],被用作光诊疗、光声治疗和光热治疗药物[115-116]、生物监测剂[117]、药物治疗[118-119]等,进一步扩大了碳点的应用范围,提升了碳点的应用潜力。
5 碳点目前所面临的问题与展望
随着科研人员对碳点的研究不断深入,碳点的实际应用也在不断扩充,碳点不仅有良好的光致发光特性,还具有良好的生物相容性和环境友好性,但是目前其制备与应用方面仍存在着一些局限性:(1)合成碳点的原料来源广泛,合成方法较多,给碳点带来了多样的性质,但同时也带来了对其微纳结构的研究难度,其发光机理与形成过程尚不明确。(2)长波长发射的碳点在生物成像、文件加密等领域有巨大的应用潜力,但是由于制备方法与对其发光机理研究的局限性,高效水溶性长波长碳点目前依然很稀缺,因此如何可控地合成高效率长波长水溶性的碳点是未来发展的方向。(3)目前的合成方法制备的碳点提纯步骤复杂、有机溶剂消耗大,即使是提纯后的碳点其精确结构(分子量、组成、晶格缺陷、杂原子占位等)也还不能完全确定。因此,开发标准化的碳点合成方法、提纯流程以及精确结构表征手段具有重要意义。(4)目前碳点在荧光探针和生物成像等方面的应用已经得到广泛的研究,碳点具有高选择性、高灵敏度、响应速度快、成本低等优点,但是其应用基本还处于实验室阶段,进一步提高其稳定性、选择性与抗干扰能力,走向临床化是下一阶段的努力方向。碳点具备其他量子点所没有的诸多优点,在催化、检测、成像等领域有着巨大的应用潜力。目前,碳点不断向着制备简单、提纯方便的方向发展,相信随着对碳点研究的不断深入,碳点面临的局限性将被一一解决。
6 结 论
碳点作为一种新型的碳纳米材料,具有优异的荧光性质、低毒性、良好的生物相容性和廉价的制备成本等特点,引起了越来越多科学家的研究兴趣,并逐渐在生物成像、生物分子检测、金属离子测定等领域崭露头角。但是,碳点也面临着一些问题,例如荧光量子效率较低,发射波长主要集中在蓝绿光区,因此,进一步优化碳点的合成方法、探究碳点的发光原理是解决这些问题的一个途径。相信随着碳点合成及修饰技术的不断完善,碳点必将会在生物化学、材料等领域展示良好的发展前景。
本文专家审稿意见及作者回复内容的下载地址:http://cjl.lightpublishing.cn/thesisDetails#10.37188/CJL.20210061.
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07 06:22:18
昆明医科大学学报(2021年8期)2021-08-13 08:59:28
潍坊学院学报(2021年2期)2021-07-22 07:59:04
中央民族大学学报(自然科学版)(2018年3期)2018-11-09 01:16:32
分析化学(2017年12期)2017-12-25 12:44:25
分析化学(2017年12期)2017-12-25 07:05:04
云南师范大学学报(自然科学版)(2015年5期)2015-12-26 12:46:16
无机化学学报(2014年4期)2014-02-28 17:31:21
无机化学学报(2014年4期)2014-02-28 17:31:09
中国洗涤用品工业(2011年6期)2011-03-20 15:38:56
