
高效阴离子交换色谱-脉冲积分安培法检测海带中的单糖、双糖和糖醛酸
2021-08-31 02:34尹大芳孙晓杰郭莹莹王联珠
食品科学 2021年16期
尹大芳,孙晓杰,郭莹莹,李 娜,田 雨,王联珠,*
(1.上海海洋大学食品学院,上海 201306;2.中国水产科学研究院黄海水产研究所,山东 青岛 266071)
海洋资源丰富,海洋生物含有丰富的糖类、蛋白质、矿物质等营养物质,相对于陆生资源具有更高的营养以及更高的开发价值[1],海藻的加工以及高值化利用成为海洋资源综合利用的重要领域[2]。海带含有海带多糖、优质蛋白质、氨基酸、纤维素等多种生物活性成分,具有利水消肿和御寒等功效。海带多糖在降血糖[3]、抗肿瘤[4]、抗氧化[5]和神经保护[6]等方面具有重要的生物学活性。但由于多糖结构复杂,其生物活性机制研究仍然是一个难题。单糖组成和含量对生理活性有极大影响[7-8],是多糖基础性和关键性的研究环节[9]。单糖是一种多羟基醛酮化合物,极性强,无发色官能团和荧光官能团,结构相似,存在同分异构体,对分离条件的要求较高。
常用的单糖定性定量方法有高效液相色谱法[10-14]、气相色谱法[15]、毛细管电泳法[16-18]和高效阴离子交换色谱法[19-21]等。高效液相色谱联合示差折光检测器和蒸发光散射检测器无需衍生,操作简便,但选择性和灵敏度差[10-12]。由于单糖没有发色基团、荧光基团以及没有挥发性,采用紫外、荧光检测器以及气相色谱检测时,常需要进行柱前柱后衍生化[14-15]。衍生化过程费时繁琐,常伴随衍生过程不彻底或生成多种衍生物,阻碍了糖类检测技术的发展[15,22-24]。毛细管电泳法灵敏度低和重复性差的特点极大限制其应用[17-18,20]。高效阴离子交换色谱-脉冲安培检测(high performance anion exchange chromatography with pulsed amperometric detection,HPAEC-PAD)法是一种强有力的分析糖类化合物的检测技术。以电化学检测器作为检测器,高选择性的阴离子交换柱作为固定相,选择最新优化的四电位检测波形,负电位清洗电极,正电位氧化并激活电极,可有效降低电极的损耗,延长电极寿命以及增加实验的重复性[25];羟基的氧化电位较低(0.1 V),所以基体干扰少,方法的重复性和准确性高[26]。NaOH溶液作为淋洗液,改变NaOH浓度,可影响溶液中分子形成氢键的能力,导致其与固定相结合能力的差异实现糖类分离。电化学检测器与梯度淋洗液的相容性,结合阴离子交换固定相的高选择性,可根据糖分子结构间的细微差别,几乎可达到所有类别的醛糖醇的分离,检出限低至10-12~10-15mol[25]。该方法具有操作简单,灵敏度、选择性、准确度高和重复性好的特点。HPAECPAD已应用于各种常规监测和研究领域,包括牛奶[27]、谷类[28]、中草药[21]、玉米淀粉[29]以及细胞培养[9]等各种材料中单糖、双糖、寡糖的检测等,但水产品中糖类种类较为复杂,应用较少。此外,针对水产品的中性糖和糖醛酸同时检测的应用较少。
因此,本实验采用Dionex ICS-3000离子色谱系统,联合脉冲安培检测器,以CarboPac PA10(250 mm×4 mm)作为糖分析柱,考察淋洗液浓度、色谱柱柱温和淋洗液流速对各组分分离的影响。采用梯度洗脱对常见的11 种糖类化合物进行分离定量,建立同时定性定量分析11 种糖类化合物的方法,利用该方法对海带多糖水解后单糖组分进行测定。
1 材料与方法
1.1 材料与试剂
样品分别采购自山东威海、辽宁大连、福建霞浦海域;11 种糖类标准物质:岩藻糖(fucose,Fuc)、葡萄糖(glucose,Glc)、氨基半乳糖(galactosamine,GalN)、氨基葡萄糖(glucosamine,GlcN)、半乳糖(galactose,Gal)、甘露糖(mannose,Man)、果糖(fructose,Fruc)、核糖(ribose,Rib)、乳糖(lactose,Lac)、半乳糖醛酸(galacturonic acid,Gal UA)和葡萄糖醛酸(glucuronic acid,Glc UA)(纯度均不小于98%),50%氢氧化钠(色谱纯)、乙酸钠(分析纯) 美国Sigma公司;实验用水均采用电阻率不低于18.2 MΩ·cm的超纯水;无水乙醇、氯仿、正丁醇、三氟乙酸均属国产分析纯。
1.2 仪器与设备
Dionex ICS-3000离子色谱系统、CarboPac PA10糖分析柱(250 mm×4 mm)、CarboPac PA10糖保护柱(50 mm×4 mm) 美国戴安公司;FDU-2110高速冷冻离心机 德国Heidolph公司;DFY-300型摇摆式高速万能粉碎机 温岭市林大机械有限公司;ZRD-A7080全自动新型鼓风干燥箱 上海智试分析仪器公司;Milli-Q超纯水设备 美国Milipore公司。
1.3 方法
1.3.1 标准溶液和淋洗液的配制
标准储备液:用超纯水将各糖类化合物标准溶液均配制成1 mg/mL的母液。取适量母液配制成一定质量浓度的混合标准溶液。
200 mmol/L NaOH淋洗液:取50% NaOH溶液10.4 mL,用超纯水定容至1 L。
500 mmol/L NaAc淋洗液:称取NaAc固体20.5 g,用超纯水溶解并定容至500 mL;淋洗液配制完成后立即通氮气摇匀备用。
1.3.2 样品前处理
多糖提取:参照文献[30]的方法。洗净海带表面泥沙等杂质,在50 ℃烘箱中烘干,打碎成海带粉,-20 ℃保存备用。1.0 g海带粉加入10 倍的无水乙醇静置12 h,取沉淀于50 ℃干燥箱中干燥至恒质量;加入30 倍超纯水,利用超声细胞粉碎机处理30 min,置于60 ℃恒温水浴锅中浸提30 min,离心取上清液,于60 ℃旋蒸至原体积的1/4,加无水乙醇至体积分数80%,4 ℃过夜,离心,取沉淀复溶于水,利用Sevag法(正丁醇-氯仿,1∶5,V/V)除蛋白后,用2 mol/L三氟乙酸溶液于120 ℃恒温干燥箱中水解2 h;水解完成后NaOH中和水解液,离心,去沉淀,经0.22 μm滤膜过滤,用于进样分析。
1.3.3 色谱条件
采用CarboPac PA10(250 mm×4 mm)作为糖分析柱,CarboPac PA10(50 mm×4 mm)作为糖保护柱,脉冲安培检测器,糖标准四电位(表1)作为检测波形,水-NaOH-NaAc组成三元淋洗液,流速为1.0 mL/min,柱温为25 ℃,进样量为25 μL。淋洗液系统最终优化洗脱程序见表2。梯度洗脱程序可分为单双糖的分离、糖醛酸的分离、基质的消除、赋予色谱系统强pH值、色谱柱的平衡5 个阶段。利用保留时间进行定性,外标法(峰面积)进行定量计算。图1为质量浓度10 mg/L混合标准溶液色谱图。
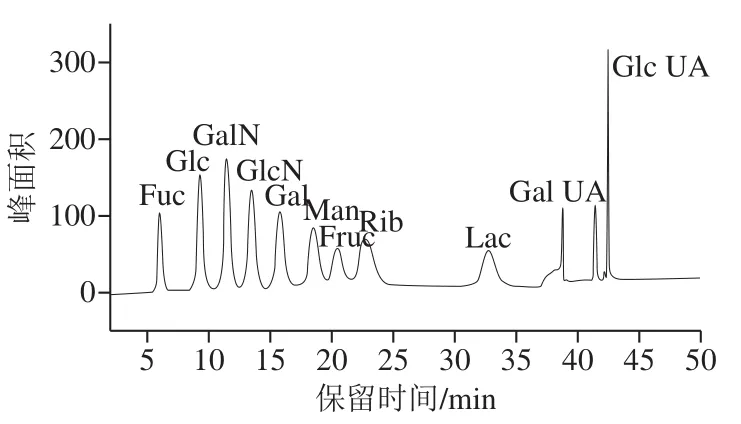
图1 11 种糖类混合标准溶液色谱图Fig. 1 Chromatogram of mixed solution of 11 sugar standards
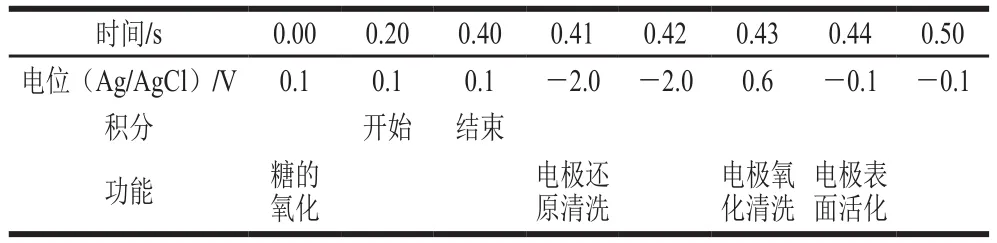
表1 糖标准四电位波形Table 1 Quadruple-potential waveform for carbohydrate detection
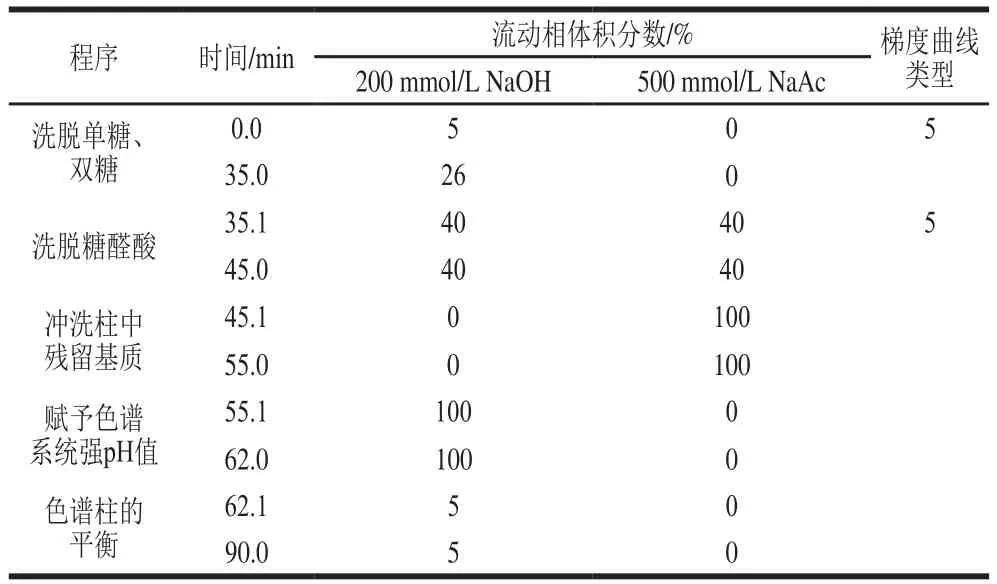
表2 梯度洗脱程序Table 2 Gradient elution program
2 结果与分析
2.1 淋洗液优化
糖类具有复杂的空间结构,对于一些差向异构体的单糖(如Glc与Gal、Man)pKa相近,易形成共洗脱,较难分离[31]。通过淋洗液的浓度改变淋洗液的pH值,从而改变糖类化合物的质子解离程度,达到有效的分离。糖类在色谱柱上保留性质与糖类的pKa值具有重要的相关性,表3为部分糖类化合物的pKa值。相同的碱性环境中,pKa值越大,其酸性越弱,糖类解离度越差,与固定相结合能力越弱,越容易洗脱;糖类在阴离子交换分离柱的洗脱顺序依次为单糖、双糖、糖醛酸。糖醛酸是糖的衍生产物,比相应的单糖多了羧酸基团,酸性较强,在阴离子交换柱上的保留性强。
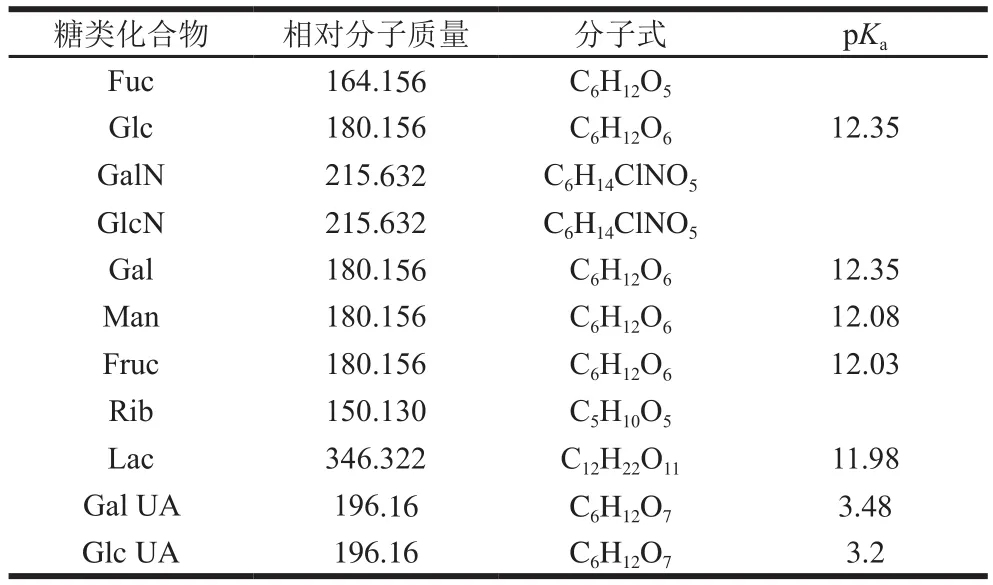
表3 部分糖类化合物的pKa值(水中,25 ℃)Table 3 pKa value of some sugars (in water at 25 ℃)
针对11 种糖类化合物的洗脱条件优化,本实验分为2 部分完成:第1部分为8 种单糖和1 种双糖(Fuc、Glc、GalN、GlcN、Gal、Man、Fruc、Rib和Lac)的洗脱优化,中性糖在色谱柱上的保留时间相对较短,其单糖结构相似,具有同分异构体,淋洗液浓度对其洗脱具有决定性作用;第2部分为糖醛酸(Glc UA和Gal UA)的洗脱优化,糖醛酸的pKa值小,偏酸性,在色谱柱上的保留时间长,NaOH溶液作为洗脱剂时,很难对糖醛酸分离洗脱;而NaAc与固定相的亲和力强,Ac-洗脱能力强于OH-,因此,使用NaAc对糖醛酸进行洗脱实验。
2.2 单糖和双糖的等度洗脱实验
2.2.1 淋洗液选择
选取5、10、20、25、30 mmol/L NaOH溶液对9 种糖类化合物进行等度洗脱,考察对色谱峰的分离度与峰形的影响,结果如图2所示。实验过程中发现:淋洗液NaOH浓度低于5 mmol/L时,色谱峰展宽,分析时间较长,洗脱时间超过60 min;淋洗液为10 mmol/L NaOH溶液时,Fuc、Glc、GalN三种单糖之间的分离度较高,峰形较好,但对于较难洗脱的组分(Gal、Man、Fruc和Rib)间的分离度较差,且洗脱时间相对较长,超过40 min。当淋洗液NaOH浓度为20 mmol/L时,各单糖之间分离较好。当淋洗液NaOH浓度继续增大时,分析时间逐渐缩短,色谱峰峰形变尖,分离度呈现下降趋势。
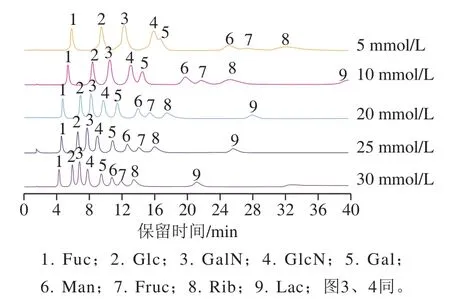
图2 NaOH浓度对9 种糖类化合物分离效果的影响Fig. 2 Influence of NaOH concentration on chromatographic separation of nine sugars
随NaOH浓度的增大,各个色谱峰保留时间缩短,响应值增高。这可能是由于NaOH浓度的变化可以改变溶液中OH-浓度、溶液pH值以及糖类在溶液中的解离度。随OH-浓度增加,糖类的解离度增大,导致在固定相上的保留时间延长;但是,随OH-浓度增加,淋洗液的洗脱能力增强,亦可使糖类在固定相上的保留时间缩短。其中,后者占据主导优势。在考虑分离度、色谱峰的峰形、分析时长等因素的情况下,选择20 mmol/L NaOH溶液作为流速与温度优化程序的固定条件。
2.2.2 流速选择
以20 mmol/L NaOH溶液作为淋洗液,设定淋洗液的流速分别为0.4、0.6、0.8、1.0、1.2 mL/min,考察流速对9 种组分的色谱峰分离度与峰形的影响,结果如图3所示。结果表明,流速低时分析时间长,峰形变宽,柱效降低,对于易洗脱的单糖(Fuc、Glc、GalN、GlcN和Gal)分离度更高,峰形更好;流速的变化对于Man、Fruc、Rib和Lac等较难洗脱的糖类化合物则影响较小;流速高时,柱压升高,各糖类化合物分离度变差。考虑分离度、色谱柱压力、色谱柱柱效及分析时长等因素,选择1.0 mL/min作为最终流速。
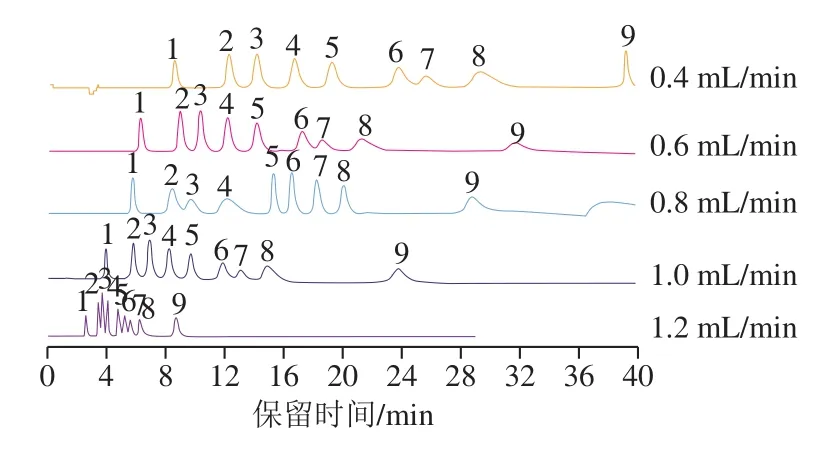
图3 流速对9 种糖类化合物分离效果的影响Fig. 3 Influence of mobile phase flow rate on chromatographic separation of nine sugars
2.2.3 温度选择
以20 mmol/L NaOH溶液作为淋洗液,流速为1.0 mL/min,改变色谱柱温度分别为20、25、30、35、40 ℃,观察各组分间分离度和峰形的变化,结果如图4所示。当柱温在25 ℃及以下时,分离度相对较好。柱温在30 ℃以上时,糖类化合物的分离度变差,且不能实现Glc和GalN的分离。
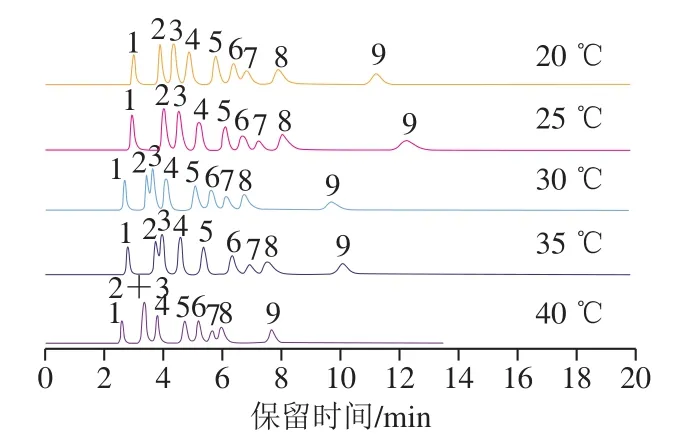
图4 温度对9 种糖类化合物分离效果的影响Fig. 4 Influence of column temperature on chromatographic separation of nine sugars
结果显示,随温度升高,色谱峰的保留时间呈减小趋势。这是由于糖类在色谱柱的保留属于放热行为,不同糖类化合物间受影响的程度不同,一般规律为随糖类化合物相对分子质量的增大,保留时间的影响程度增大[32]。柱温对HPAEC分离单糖具有重要的影响,很小的温差变化(±5 ℃)便会引起单糖保留时间的偏移,影响方法的重复性。考虑到分离度、峰形以及仪器耐受性等因素,选择25 ℃作为色谱柱温度。
2.3 单糖、双糖和糖醛酸的梯度淋洗液系统的确定
Fuc、Glc、GalN和Gal四种糖类化合物的保留时间较短,低浓度的NaOH溶液可达到较好的分离效果。Man、Fruc、Rib和Lac在固定相中的保留性较强,需采用较高浓度的NaOH溶液进行洗脱分离。但洗脱过程中不可遽然改变淋洗液浓度,否则基线漂移严重,影响后续定量。采用在0~35 min内,10~52 mmol/L NaOH作为梯度洗脱实现对单糖和双糖的洗脱。糖醛酸在色谱柱上保留性强,若单独使用200 mmol/L NaOH进行洗脱,洗脱时间长达60 min。需加入强洗脱剂NaAc溶液(与色谱柱亲和度更高)作为淋洗液,最终确定以200 mmol/L NaAc+80 mmol/L NaOH对糖醛酸进行洗脱。在完成检测过程后,采用与固定相亲和力更强的NaAc对保留性更强的杂质进行清除,本实验采用500 mmol/L NaAc作为基质消除洗脱液。淋洗液系统的最终优化洗脱程序见表2,总运行时间为90 min。
2.4 方法学验证
2.4.1 标准曲线
分别配制质量浓度为0.5、1.0、2.0、5.0、10.0、20.0、40.0、60.0 mg/L的11 种糖类化合物的混合标准溶液,以质量浓度(x,mg/L)为横坐标,以峰面积(y)为纵坐标,绘制标准曲线。各组分的保留时间、线性关系、线性范围、相关系数、检出限(RSN=3)、定量限(RSN=10)见表4。11 种糖类化合物在各自线性范围内线性关系R2不小于0.998 6,方法的检出限(RSN=3)在1.27~47.49 mg/kg之间。

表4 线性数据和检出限Table 4 Linearity and limits of detection and quantification
2.4.2 精密度结果
对日内精密度和日间精密度进行考察,分别从保留时间和峰面积2 个方面进行测定。在优化的色谱条件下,对质量浓度10 mg/L的混合标准溶液连续进样7 次计算日内精密度,各单糖保留时间的相对标准偏差(relative standard deviation,RSD)均小于0.259%,峰面积的RSD均小于1.69%。将10 mg/L的混合标准溶液1 d进样3 次,连续进样4 d,计算日间精密度。各单糖保留时间的RSD均小于0.27%,峰面积的RSD均小于1.78%。结果见表5。
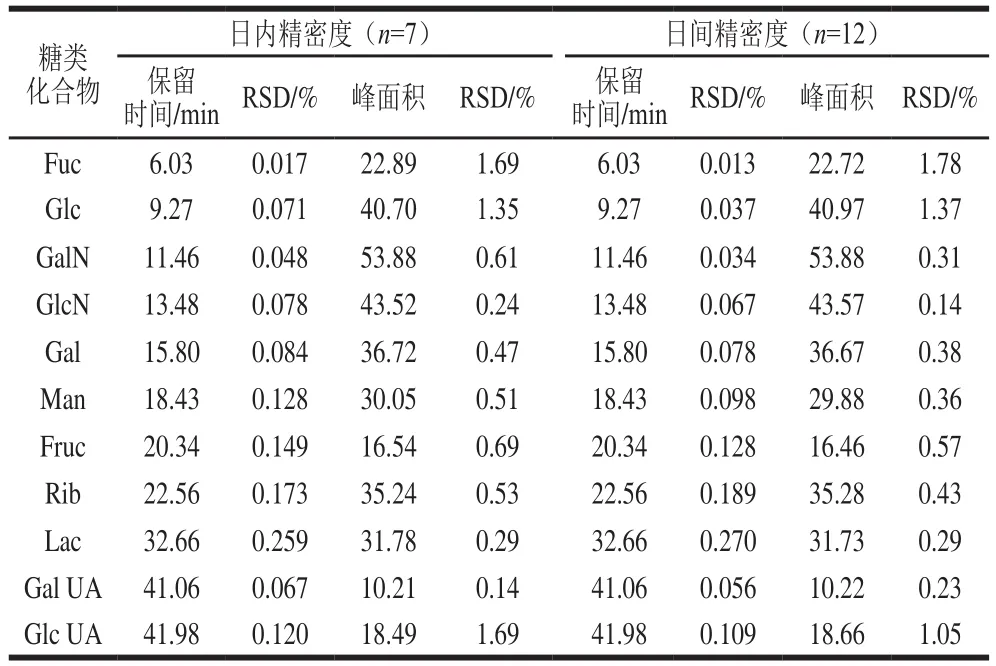
表5 精密度实验结果Table 5 Results of precision experiments
2.4.3 加标回收率结果
对海带多糖的水解液进行加标回收率实验,对于样品中含有的单糖,以样品中单糖质量分数的50%、100%、200%进行加标,对于样品中不含的糖类,以1、2、4 mg/L进行加标实验,重复进样7 次,计算峰面积的RSD。由表6可知,样品加标回收率在89.35%~115.67%之间。
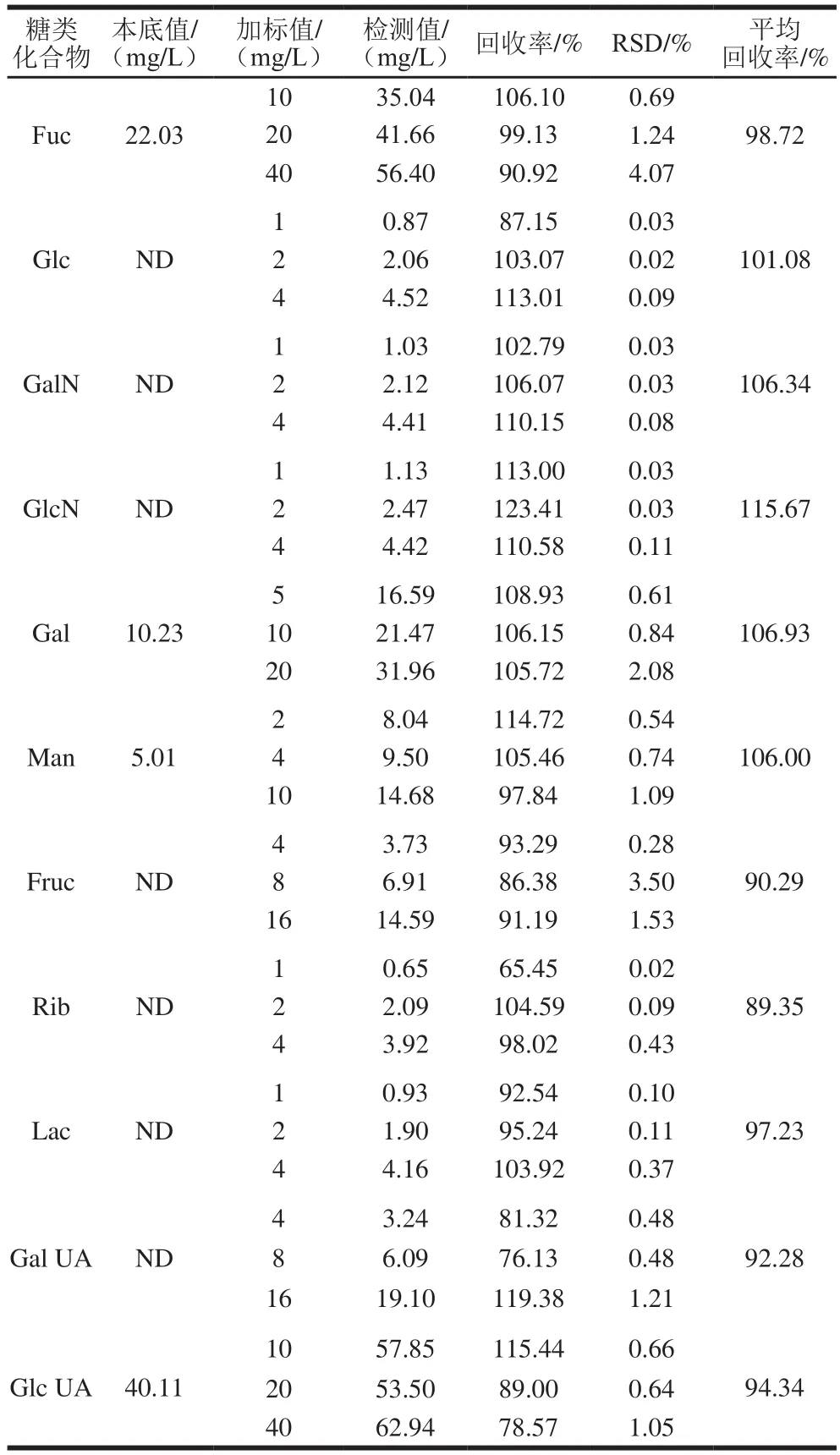
表6 加标回收率实验结果(n=7)Table 6 Results of spiked recovery experiments (n = 7)
2.5 样品的测定
采用1.3.2节方法对8 份海带样品进行前处理,采用1.3.3节色谱条件进样分析,其中山东威海海之宝海域的1号样品及其样品加标色谱图见图5。每批样品平行测定3 次,取平均值,得到海带样品的糖类组成和含量,结果见表7。
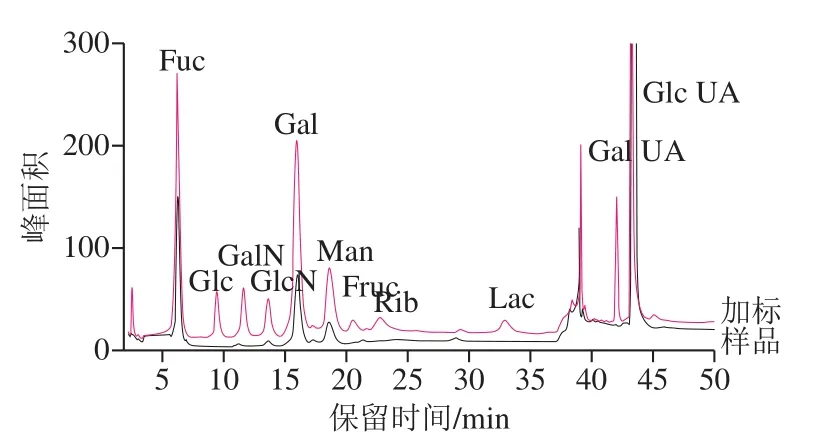
图5 海带样品及其加标色谱图Fig. 5 Chromatograms of polysaccharides in real and spiked samples of Laminaria japonica
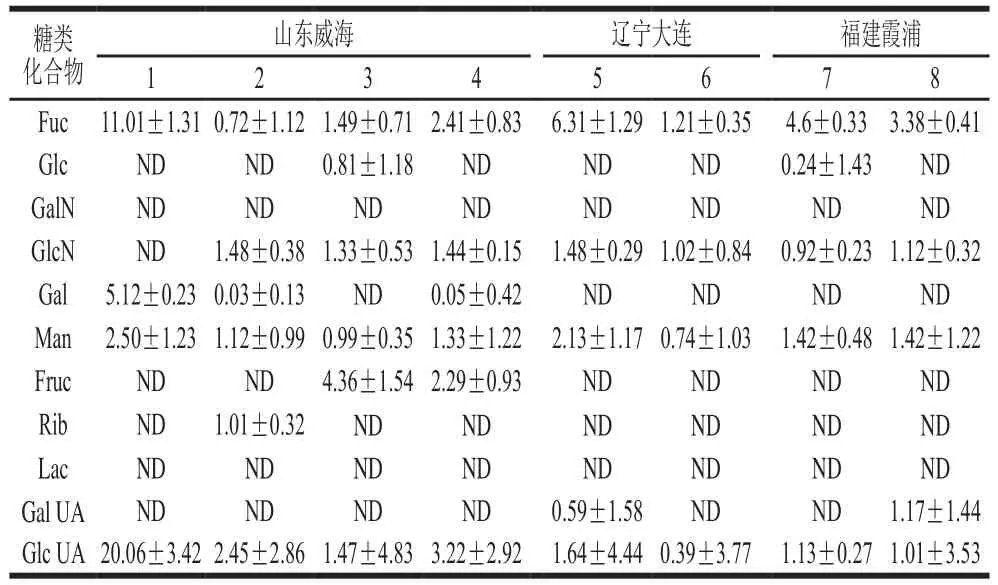
表7 实际样品糖类化合物含量(n=3)Table 7 Contents of sugars in actual samples determined by the developed method (n = 3) mg/g
3 讨论与结论
水产品中多糖结构复杂,单糖种类多样,单糖间分离难度较大。色谱柱的选择是影响单糖分离的重要因素,CarboPac PA1、CarboPac PA10以及CarboPac PA20阴离子交换柱常用来分离单糖、双糖。CarboPac PA1分析糖类时具有明显的溶解氧负峰,且CarboPac PA1的灵敏度较CarboPac PA10和CarboPac PA20低[25];CarboPac PA10和CarboPac PA20色谱柱的填料大致相似,不同的是CarboPac PA10色谱柱树脂基核及覆盖在树脂表面的键合是由双功能乳胶构成,而CarboPac PA20则是由季铵功能基乳胶构成,CarboPac PA10容量相对更大[33]。本实验对中性糖和糖醛酸同时进行分离,需采用较大容量的色谱柱。CarboPac PA10由乙基乙烯基苯-二乙烯基苯聚合形成的基核,表面附聚具有疏水性的双功能乳胶颗粒,容量为100 mol/柱,对溶解氧的保留性更强,可有效避免氧负峰[34]。最终选取CarboPac PA10作为11 种糖类组分分析的阴离子色谱柱。
本研究建立梯度洗脱-HPAEC-PAD分析Fuc、Glc、GalN、GlcN、Gal、Man、Fruc、Rib、Lac、Glc UA和Gal UA 11 种糖类化合物的检测方法,实现了对中性糖、氨基糖和糖醛酸的同步分离,且11 种糖均达到基线分离,线性关系良好,精密度高。对海带多糖水解液进行加标回收率实验,其加标回收率在89.35%~115.67%之间。相对于其他检测方法(如高效液相色谱法)可分析的单糖种类来看,HPAEC-PAD可检测的种类更多,更全面,分离度更高。相对于气相色谱等其他需衍生化的检测方法看,操作更为简便、高效。结果表明,该方法具有操作简便易行,分离度、灵敏度、精密度高,重复性好等特点。
猜你喜欢
第一财经(2020年10期)2020-10-15
第一财经(2020年3期)2020-02-29
中成药(2019年12期)2020-01-04
第一财经(2019年11期)2019-12-02
第一财经(2019年7期)2019-07-25
天然产物研究与开发(2019年1期)2019-03-01
天然产物研究与开发(2018年6期)2018-07-09
家庭用药(2018年11期)2018-01-23
中国民族医药杂志(2016年7期)2016-05-09
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
