
分子模拟在油气地球化学中的应用研究进展
2021-08-30 06:47李美俊刘晓强韩秋雅方镕慧何大祥高志伟
石油与天然气地质 2021年4期
李美俊,刘晓强,韩秋雅,肖 洪,方镕慧,何大祥,高志伟
[1.页岩油气富集机理与有效开发国家重点实验室,北京 102206; 2.中国石化油气成藏重点实验室,江苏 无锡 214126;3.中国石化 石油勘探开发研究院 无锡石油地质研究所,江苏 无锡 214216; 4.中国石油大学(北京)地球科学学院,北京 102249; 5.四川轻化工大学 化学与环境工程学院,四川 自贡 643000; 6.中国地质调查局非常规油气地质重点实验室,北京100083; 7.长江大学 油气资源与勘探技术教育部重点实验室,湖北 武汉 430100]
分子模拟就是采用计算机辅助实验技术,以统计力学和量子力学为基础,基于原子水平的分子模型来模拟计算分子的结构与行为,以及体系的各种物理化学性质[1-2]。分子模拟的主要作用包括模拟物质的结构、计算物质的性质、预测物质的行为、验证实验结果以及重现实验过程等。其优势在于成本低、安全性高、具有预测性、能从原子和分子水平上直观再现物质的微观性质和各种物理化学过程等。因此,分子模拟已成为连接微观与宏观尺度的重要桥梁,为体系微观机制的探索提供分子、原子水平上的认识。
广义的分子模拟主要分为两个方面:一方面是以量子力学为主的分子模拟,即所谓的量化计算,主要为分子轨道计算,包括从头计算、半经验计算和密度泛函理论(density functional theory, DFT)计算,其研究体系往往较小,只能处理数百个原子体系,并且计算速度慢,主要依赖于算法,但在定性描述方面极具优势;另一方面是以经典牛顿力学为主的分子模拟,其在计算化学领域通常被默认为分子模拟。分子模拟主要分为分子力学(molecular mechanics, MM)、蒙特卡罗(Monte Carlo, MC)以及分子动力学(molecular dyna mics, MD)模拟。这类方法的优点是能够定性描述分子的运动轨迹,研究体系较大,可达上万个原子,计算速度快,主要依赖于分子力场,然而缺点是仅能研究系统短时间范围内的运动,而无法模拟一些时间较长的运动问题[1]。
作为一种重要的研究手段,分子模拟在石油化工、勘探、开发等生产和研究中已备受关注,并取得了一系列研究成果[3]。本文主要总结和评述分子模拟技术在油气地球化学领域的应用及进展,主要包括在煤层气吸附机理、页岩气和页岩油吸附、有机质热成熟度评价的分子地球化学指标建立、油气运移方向地球化学示踪、有机质生烃和油藏中原油裂解机理等油气地球化学重点关注的研究领域,并简述其应用前景及未来发展方向。
1 煤层气吸附机理
分子模拟在化学化工和材料科学等领域的应用较为成熟。地质体中岩石、煤和有机质等为含有类似于微孔的天然材料,而地下的油、气、水同样是复杂的溶液和流体,因而,化学化工和材料科学领域的分子模拟成果同样对煤层气/页岩气吸附和扩散微观机理、成藏规律及开发生产具有一定借鉴意义。
关于煤对天然气的吸附,已有许多研究成果相继报道了CH4,CO2,N2和H2O等天然气组分在煤结构模型上的吸附机理,获取了诸如吸附构型、吸附位点、吸附能、电子转移和官能团效应等物理化学特征[4-5],对深入探索煤层气在煤中的吸附机理提供了定性认识。为了深入了解煤层气在煤的大分子三维网络结构中的吸附行为,通常采用分子模拟技术探索煤与瓦斯之间的微观吸附机理,并直观、准确地对宏观体系进行定量认识。例如,Mosher等(2012)采用巨正则蒙特卡罗(GCMC)模拟方法,预测了CH4在0.4~9 nm孔径的碳分子狭缝中的等温吸附特征[6]。Liu等(2012)的研究结果发现,煤分子结构中的含氧官能团对CO2吸附具有很重要的影响[7]。Zhang等(2014)采用GCMC和MD模拟方法,研究了CH4在干燥和含水煤分子的等温吸附[8]。分子模拟结果表明,在相同的温度和压力下,煤对CO2的吸附量大于CH4,因此可以在煤层气开采中采用注入CO2的方式提高煤层气的采收率,即所谓CO2-ECBM技术,同时还可以封存CO2以减少温室气体的排放[9-13]。
事实上,煤层气在不同煤结构中的吸附模拟已取得了不少进展和突破,但如何将分子模拟与地质研究有机结合、如何将理论研究和计算成果应用于实际的煤层气勘探实践,还有很大的努力空间。近年来,本研究团队采用分子动力学模拟技术探索了煤层气在不同地质埋深煤层中的微观吸附机制,深入研究了压力与温度的耦合效应。此外,还从理论上首次提出吸附百分比(δ)和采收率(η)两个重要参数,预测了在典型地质条件下,实际开采过程中CO2促进CH4采收的最佳开采深度约为800 m(图1)[10]。因此,本团队提出的CO2-ECBM最佳操作条件的方法,可以从分子尺度上深入了解不同埋深煤样中气体分子吸附的行为,为煤与瓦斯共采提供较深入的理论认识。同时,还可以揭示地质埋深对实际生产中煤层气或页岩气驱替和CO2封存的微观机制。根据理论研究和实验结果,该方法还可应用于致密油气藏、超深层天然气、页岩气和页岩油的扩散、聚集和运移等微观机理。总之,未来煤与瓦斯共采依然是重点攻克的关键性难题,理论模型与地质条件充分结合是必然之路,分子模拟作为实验及工程的辅助手段必大放异彩。
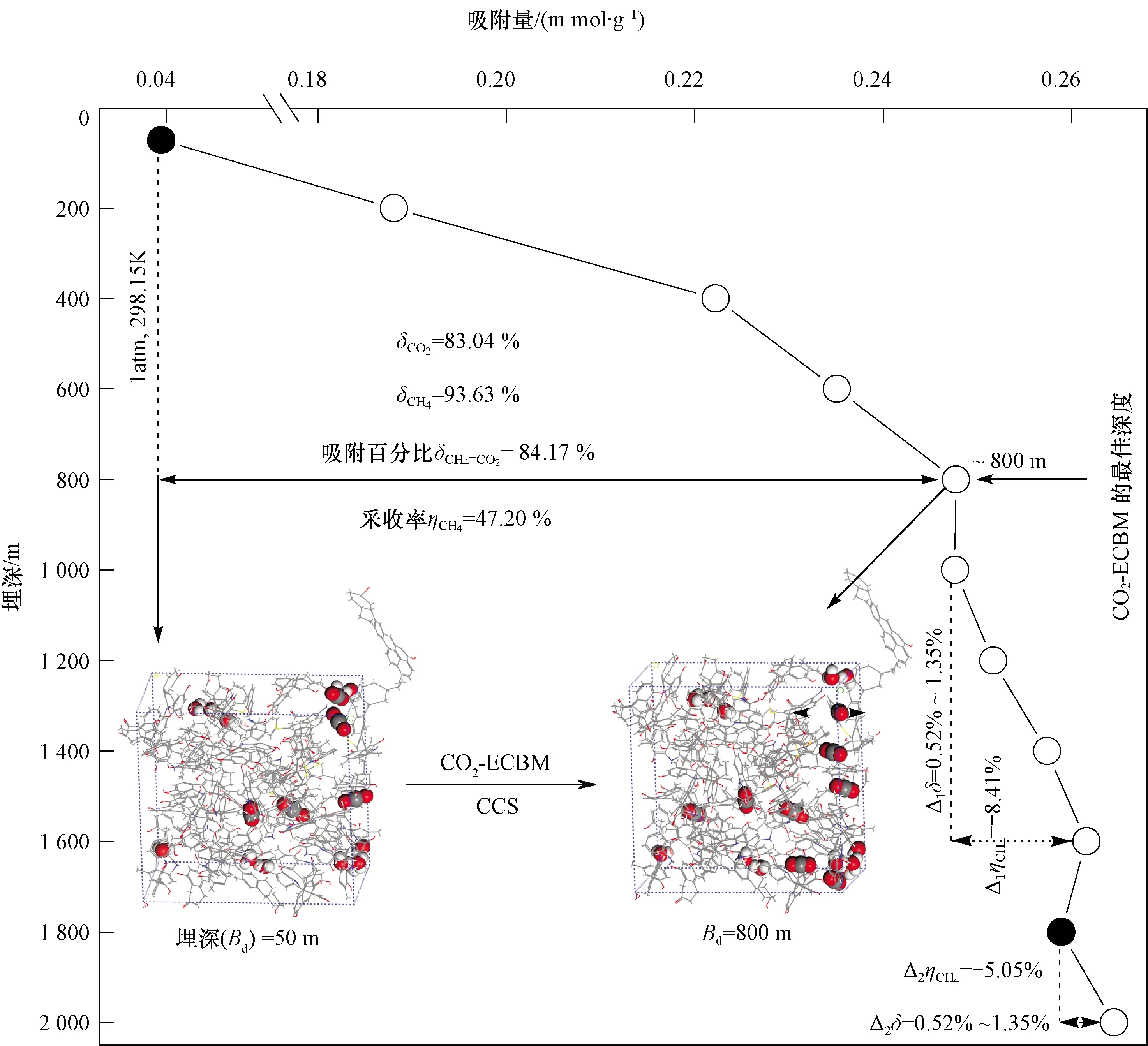
图1 实际开发过程中CO2-ECBM的最佳开采深度预测[10]
2 页岩气/页岩油吸附机理
页岩气是重要的非常规油气资源,全球页岩气资源量约为456×1012m3[14],中国页岩气资源丰富,可采资源量可达15×1012~25×1012m3[15]。不同于常规天然气,页岩气一个显著的特点是吸附气含量高,介于20%~85%,一般在50%左右,约占页岩气总产量的40%,因此页岩气吸附机理研究与吸附量计算对页岩气资源评价和勘探开发非常重要。
前人研究页岩气吸附规律主要通过室内实验(物理模拟实验)获取页岩气在不同条件下的吸附量以及页岩储层条件、有机质性质和水分等对气体吸附量、吸附热及吸附规律的影响[16-18]。这些实验研究取得了不少有价值的成果,对指导页岩气中的吸附气含量计算、影响因素和变化规律研究具有重要的实践意义。室内实验的优势在于:可以选取实际的地质样品开展实验,得出的结果对研究区具有直接的应用价值,并且实验结果和数据直观可见。
同时,室内实验也存在明显的不足,而这些不足之处正是分子模拟技术的优势。
1)成本高。目前的实验多是单组分吸附实验,比如CH4和CO2等,对混合组分开展的实验研究不多,只有少部分成果涉及到双组分,或者三组分混合气体的实验模拟。以天然气的等温吸附实验为例,每改变一种气体组分或者不同配比的混合气体,就需要按照完整的流程重新做一次实验,费时、费力、成本高。然而,分子模拟可以选取任意比例的混合气体进行模拟计算,可以快速、低成本地获得较高价值的研究认识。
2)受实验条件的限制。实验仪器设备模拟的温度和压力条件不能覆盖地质温度和压力范围[19],特别是高温高压条件下,页岩气吸附机理及变化规律的认识明显受限,而且在这种高温高压条件下进行实验,也会带来很大的实验风险和安全隐患。
3)实验室模拟的局限性。实验室(物理模拟)结果往往局限于关注气体吸附量,不能系统深入地揭示页岩气微观吸附机理。而分子模拟技术却可以获得气体密度分布曲线、吸附层数、吸附热、结合能和孔径效应等反应吸附特性的参数,进而深入揭示页岩气吸附的微观机理。
早期的页岩气分子模拟,主要采用GCMC和DFT计算方法,基于一些简化的吸附质模型,例如石墨狭缝模型、碳纳米管等,多为带有不同官能团的理想碳材料。近些年来,学者们开始关注天然气与页岩中不同岩石矿物之间的吸附机理研究。例如,卢双舫等模拟了不同孔径的伊利石狭缝型孔隙中页岩气的吸附行为,结果表明CO2的吸附能力强于CH4,它们与伊利石的表面吸附并非严格的单分子层吸附,理论与实验模拟结果一致[19]。
干酪根为沉积岩中的分散有机质,尽管在页岩总体积和总质量中仅占很小比例,但由于其巨大的比表面积和对气体分子较强的亲和力,干酪根成为页岩吸附气的重要赋存空间。因此,以干酪根作为吸附剂开展天然气的吸附行为,对研究页岩气吸附、成藏、资源评价和开发都具有重要的意义。近些年来,人们开始采用分子模拟技术,研究页岩有机质与天然气的吸附和扩散机理。采用GCMC和MD方法模拟了页岩气不同混合组分在干酪根有机纳米孔隙结构中的吸附机制、扩散机理以及吸附热等物化参数[20-21]。得出了一些基本认识,比如CH4和CO2在Ⅱ型干酪根中的吸附符合Langmuir定律、CO2比CH4的吸附量大、干酪根中S原子与CH4和CO2有较强的相互作用等。总的来看,干酪根对页岩气的吸附特征与煤基本类似。
在页岩油吸附和成藏方面,王森等(2015)以6层石墨烯代表有机质壁面,以正庚烷代表原油,基于OPLS力场,采用粗粒子模型,利用MD方法模拟了页岩有机质孔缝内液态烃的赋存状态。结果发现在油藏条件下,有机质孔隙内液体发生多层吸附,烷烃的密度分布不均匀,靠近固体壁面处的烷烃形成“类固体层”,其密度约为游离态液体密度的1.9~2.7倍[22]。张海波(2017)以辛烷代表页岩油分子,模拟了原油在不同有机质含量页岩中的吸附和扩散特征[23]。Webb等也模拟了正十六烷液态烃在硅沸石表面的界面性质和吸附特征[24]。以上这些简单的模拟计算为进一步研究页岩油的吸附机理提供了重要的参考[25]。
分子动力学模拟在页岩气和页岩油的研究中刚刚起步,地质体中的矿物、有机质、原油分子模型以及大规模的计算方法有待进一步开发和完善,未来的研究将着重考虑以下4个方面:①建立接近于不同类型有机质的干酪根分子模拟;②根据实际岩石样品分析数据,有针对性地建立接近研究区典型岩石组成和结构的分子模型;③以实测的原油和天然气样品化学组成分析结果为依据,建立接近于真实的原油分子模拟;④还需要考虑多种混合流体的耦合作用、流体相态的变化等。总之,实验研究、理论分析与分子模拟技术相结合的综合研究方法,将是探索页岩气和页岩油地球化学、资源评价和成藏规律等最有效的研究手段。
3 用于构建热成熟度的分子地球化学指标
与分子标志化合物相关的成熟度指标是评价有机质和原油热演化程度的常用参数。早期成熟度参数的建立主要基于理论分析和实际的地球化学研究成果。例如油气地球化学中常用的C31升藿烷差向异构体成熟度参数[22S/(22S+22R)]、C29规则甾烷异构化成熟度参数[20S/(20S+20R)]等,其主要原理是生物体中的前驱物只存在特有的构型,在埋藏热演化过程中逐渐转化为地质构型和生物构型的混合物[26],与实际地质样品热成熟度的对比研究,可以确定分子地球化学指标与成熟度(例如镜质体反射率Ro,%)的关系。
Ts/(Ts+Tm)是常用的成熟度参数,其主要原理是,在后生作用阶段Ts[18α(H)-22,29,30-三降藿烷]稳定性比Tm[17α(H)-22,29,30-三降藿烷]的热稳定性高[26]。图2为Ts和Tm的化学结构和构象,Ts与Tm的化学结构差别在于前者的一个甲基位于C-17位(图2b),而Tm相应的甲基位于C-18位(图2a)。从立体化学考虑,Tm化合物C-18甲基的空间位阻效应明显高于Ts(图2),造成其热稳定性相对偏低。该成熟度指标也得到了许多油气化学研究实例的证实[27]。
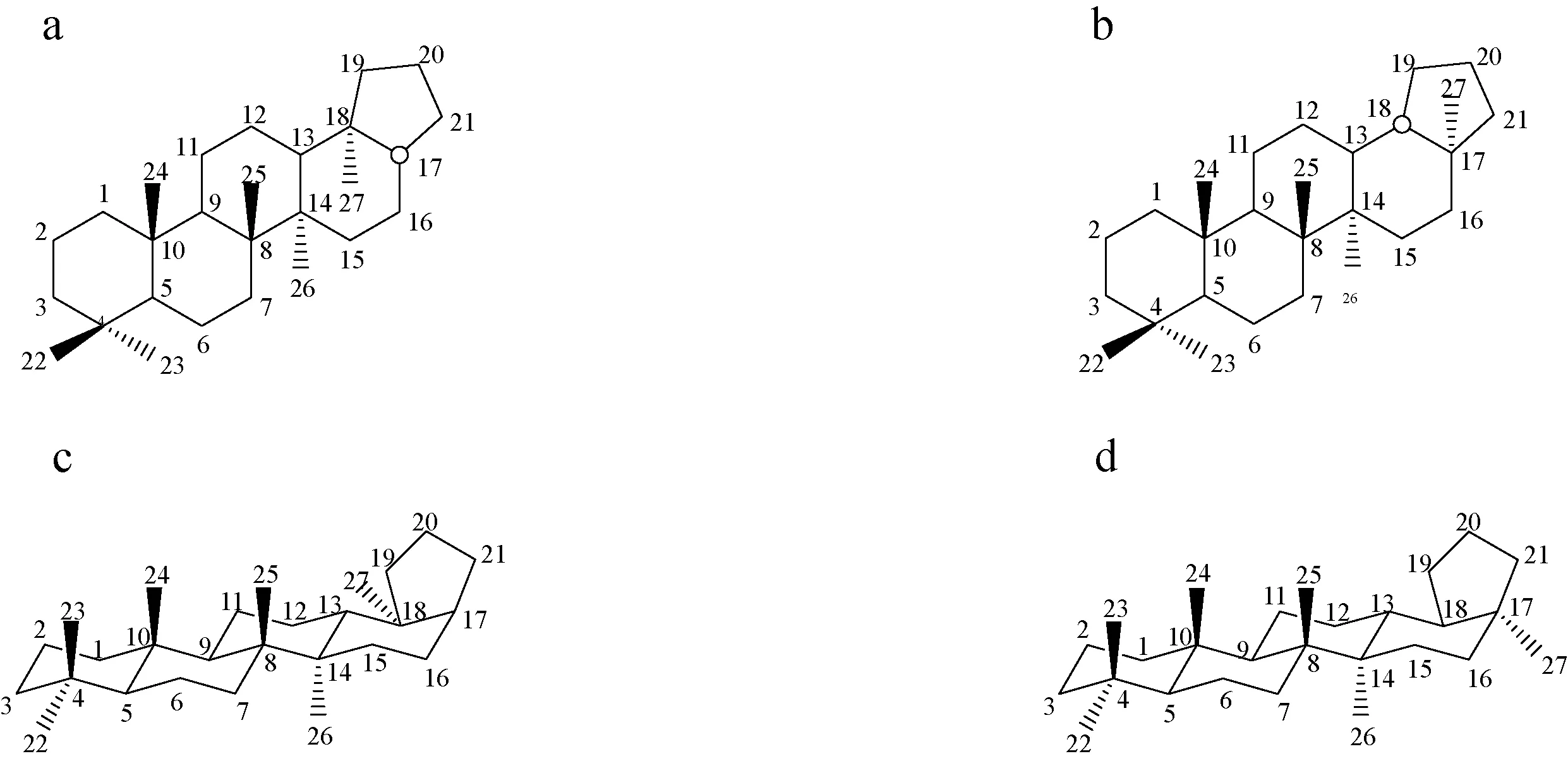
图2 Ts和Tm的化学结构和构象
Kolaczkowska等(1990)应用分子力学对包括Ts和Tm在内的6种藿烷的热稳定进行了计算,结果与理论推导和地质发现一致[28]。分子模拟为相关成熟度参数的构建提供了更坚实的证据。
此外,例如常见的与芳烃类化合物相关的成熟度参数——甲基菲指数[例如MPI-1,定义为1.5×(2-甲基菲+3-甲基菲)/(菲+1-甲基菲+9-甲基菲)][29],其理论依据在于菲环上β位取代的甲基菲异构体(例如2-甲基菲、3-甲基菲)比α位取代的异构体(例如1-甲基菲、9-甲基菲)热稳定性相对高一些,因此随着热成熟度增加,热稳定性较高的异构体含量相对增加[29],当成熟度达到Ro=1.35%以后,出现倒转[26,30]。在实际应用过程中,研究人员也发现了菲和甲基菲的相对含量影响因素复杂[31],例如沉积环境的影响[32-33]。多环芳烃与其对应的烷基取代同系物之间在地质条件下会进行复杂的相互作用、生成作用、分解破坏、去甲基化、甲基迁移和甲基取代等[34-35]。Szczerba采用分子模拟(密度泛函理论)方法并结合实测地球化学数据,计算了菲和各甲基菲异构体的热稳定性,以及甲基迁移、甲基化和去甲基化的反应过渡态和能垒,从而得出结论:常用的甲基菲指数MPI-1不完全反映热成熟度的作用,是烃源岩有机质复杂过程的综合体现,并且也受地层中矿物基质催化作用的影响,并且指出基于Ⅲ型干酪根的MPI-1-Ro关系式在成熟度大于1.35% 时,并不发生倒转[34]。因此,分子模拟为油气地球化学研究提供了更深入、更可靠的证据,有助于研究人员更科学地使用这些参数进行相应的油气地球化学研究工作。
另外一个实例是关于甲基二苯并噻吩成熟度参数的构建。前人提出了甲基二苯并噻吩(MDBT)相关的成熟度参数[4-/1-MDBT,定义为芳烃色谱-质谱(m/z=198)图中4-甲基二苯并噻吩与1-甲基二苯并噻吩化合物峰面积的比值],Radke将其归因于1-甲基二苯并噻吩在异构体中的热稳定性最低。Budzinski等提出甲基二苯并噻吩同系物的热稳定性降序为:4-MDBT>2-MDBT>3-MDBT>1-MDBT[36]。Richard计算了不同异构体的生成焓,得出4-MDBT的生成焓是23 755 kcal/mol,比1-MDBT低5 979 kcal/mol,即4-MDBT的热稳定性最高[37]。
本研究团队最新的研究成果采用分子模拟方法计算了甲基二苯并噻吩的电子能、内能、焓以及吉布斯自由能等热力学参数,并采用密度泛函理论计算了地质体中甲基二苯并噻吩甲基化、去甲基化和甲基迁移的可能途径和能垒(图3)。结果表明,酸催化作用下的1,2-甲基迁移是生油窗早期阶段(<80 ℃)影响甲基二苯并噻吩异构体相对含量的主要反应过程,4-MDBT更容易形成,加上4-甲基二苯并噻吩较高的热稳定性,导致了4-/1-MDBT随成熟度增加而升高。在成熟-高成熟阶段(<160 ℃),以自由基作用的甲基化反应机理为主,同时发现,由于较高温度下的热降解作用,导致各个MDBT异构体的绝对含量都在降低,由于1-MDBT的热稳定性相对较低,其含量下降更快,从而导致4-/1-MDBT比值也随成熟度增大而增加[35],并不是以前推测的不稳定的1-MDBT含量降低,而最稳定的4-DMBT含量升高这样一个简单认识。分子模拟研究结果为该成熟度参数的建立提供了更可靠的依据和内在的化学机理。

图3 二苯并噻吩与甲基二苯并噻吩甲基迁移、甲基化和去甲基化反应路径示意图[35]
近几年来,越来越多的研究开始采用分子模拟手段,研究苯基菲[38]、三联苯[39]以及苯基二苯并噻吩[40]等异构体的热稳定性,并构建了相应的成熟度参数。例如本研究团队采用标样共注的方法,首次在煤中检测和鉴定了苯基二苯并噻吩(PhDBT)系列化合物,并采用分子模拟方法,计算出了各异构体的热力学性质,包括吉布斯自由能、焓、电子能量和内能等,确定了该系列的热稳定性顺序:3-PhDBT>2-PhDBT>4-PhDBT>1-PhDBT,首次提出了相关成熟度参数——苯基二苯并噻吩比值[PhDR1=2-/4-PhDBT;PhDR2=(2+3)-/4-PhDBT];根据实测的地球化学数据,初步建立了当Ro大于1.00%时,苯基二苯并噻吩比值与镜质体反射率的关系式(Rc为计算得到的镜质体反射率值):
Rc(%)=0.60PhDR1+1.00
(1)
Rc(%)=0.30PhDR2+1.00
(2)
同时,发现沉积环境对苯基二苯并噻吩比值影响很小,认为该参数是定量评价高成熟原油和烃源岩(Ⅱ和Ⅲ型干酪根)成熟度的良好指标[40]。该参数为深层-超深层高演化的烃源岩和原油的成熟度评价提供了有效的分子地球化学指标。
4 油气运移分子地球化学
基于成熟度梯度和油气运移地色层分馏效应原理,油藏地球化学研究可示踪油气运移方向、优势充注路径和潜在烃源灶方位,从而定位"卫星"油气藏,直接指导油气勘探工作[41-44]。咔唑、苯并咔唑及其烷基取代同系物是石油中主要的中性含氮化合物,是油藏地球化学研究中广泛应用的油气运移方向和充注途径示踪分子标志物,其主要原理在于咔唑类分子的极性和氢键作用造成该类分子在地下原油运移过程产生运移分馏效应,咔唑分子上取代基位置不同的异构体,其极性和氢键作用大小的差异也造成不同异构体分子与输导层介质作用力的差异,因而异构体相对含量的大小和咔唑类含氮化合物总量的变化可以用来指示油气运移的方向和充注途径。分子动力学模拟结果表明,苯并[a]咔唑相较于苯并[c]咔唑更容易从油相中运移到水相中[45],这也解释了为什么苯并[a]咔唑/苯并[c]咔唑随运移距离增加而减少[46]。本团队最新的研究成果采用分子模拟方法,计算了甲基咔唑和部分二甲基咔唑异构体与石英矿物的作用能大小,结果表明4-甲基咔唑(4-MCA)的作用能大于1-MCA,2,5-二甲基咔唑(2,5-DMCA)和2,4-DMCA都高于1,8-DMCA。据此构建了相应的油气运移方向示踪参数,并在北部湾盆地福山凹陷古近系砂岩油藏中得到成功应用[47]。
由于硫(S)和氧(O)原子的电负性和孤对电子以及与咔唑相似的化学结构,石油中的烷基二苯并噻吩(DBTs)[48-50]和烷基二苯并呋喃(DBFs)类杂原子多环芳烃[51-53]同样具有运移分馏效应,同样是有效的石油运移方向和充注途径示踪分子标志物。而且这两类化合物具有含量高、抗生物降解能力强、分析简便和适用油藏类型多等优势,因而是具有广泛应用前景的油气运移示踪分子标志物[49]。
早期的研究工作主要基于化学结构,从理论上论证,并结合地质实例研究。例如1-甲基二苯并呋喃(1-MDBF)的甲基位于呋喃环C-1位,氧原子裸露,可称为氧原子暴露异构体(OEIs),而4-MDBF的甲基位于C-4位,氧原子被屏蔽,称为氧原子屏蔽异构体(OMIs)(图4),理论上1-MDBF极性大一些,与输导层介质的相互作用强一些,随着运移距离增加,1-MDBF相对4-MDBF含量减小,因而可以作为石油运移方向的分子标志物[51]。

图4 咔唑(a)、二苯并噻吩(b)和二苯并呋喃(c)的分子结构
近几年来,研究者逐渐采用分子模拟方法,计算了DBTs和DBFs系列化合物的化学结构、性质及其与输导层介质的作用。例如杨禄等(2018)采用简单的Connolly分子表面计算方法,模拟计算了4-MDBT和1-MDBT与水分子的吸附作用差异,论证了石油中甲基二苯并噻吩在油气运移中产生“地色层分馏效应”的机理[50]。Li等(2018)计算了二苯并呋喃和甲基二苯并呋喃(MDBFs)分子极性大小,以及与简单的无定型二氧化硅分子表面吸附能的大小,发现1-MDBF的极性远远大于4-MDBF,1-MDBF的吸附能也高于4-MDBF,因此构建了新的石油运移方向示踪分子指标1-/4-MDBF,并在南海北部边缘北部湾盆地福山凹陷古近系砂岩油藏地球化学研究中得到成功应用[52]。王宁等(2020)进一步计算了MDBF异构体与白云石之间的吸附能大小(图5),发现1-MDBF的吸附能同样高于4-MDBF,并应用1-/4-MDBF参数等值线图,成功示踪了四川盆地磨溪-高石梯地区新元古界-古生界古油藏原油的运移方向[53]。并且发现该类化合物非常稳定,在高-过成熟原油和沥青中都能以相当的浓度稳定存在,而且受成熟度影响小,MDBF上的甲基也较稳定不宜分解[53],因此是一类潜在的适用于高-过成熟油藏的分子地球化学指标。

图5 DBF,1-MDBF和4-MDBF在白云石(104)表面的吸附以及对应的吸附能(据Wang等,2020)[53]
5 有机质生烃机理
烃源岩有机质生烃机理一直是油气地球化学关注的重要基础研究领域,前人主要采用实验模拟方法结合生烃动力学计算,对干酪根的生烃特征进行研究,取得了许多有益的认识[54-55]。对研究干酪根生烃动力学机制、计算反应速率、计算生烃量和确定生烃演化历史等提供了有用的实验与理论证据。但对反应过程、特别是反应的化学机理却不能提供更多有效的信息。作为一种有效的研究方法,分子模拟在揭示原油裂解和有机质生烃机理方面有望发挥重要的作用,但目前这方面的成果还很少。
干酪根生烃机理分子模拟的关键在于构建可靠的干酪根分子模型和选择合适的反应力场。干酪根是指沉积岩石中不溶于有机溶剂、非氧化性酸的分散有机质的总称[56-57]。它是多种大分子有机化合物组成的混合物,分子结构非常复杂,也没有固定的化学式和分子结构[56-57],所以不可能构建干酪根分子模型。为了研究方便,人们提出了“平均分子结构”的概念。“平均分子结构”是指将结构中的芳簇数进行平均后的原子集合体,不能把它当作全体中任何一个具体分子的化学结构或者分子的基本单元,而是将其视为数学模型的一种,并借此反映混合物分子群体的典型化学与物理性质[57],这种方式已经在煤、油页岩有机质化学结构的研究中被广泛使用[58]。
Ungerer等(2014)据Kekemen等人的分析数据构建了6种不同类型和成熟度的干酪根模型,并分析了它们的体积和热物理性质[59]。由于世界范围内页岩气资源勘探的兴起,越来越多的研究开始关注页岩气在干酪根中的吸附机理分子模拟研究,这6种经典的干酪根模型被后来的研究者直接采用[20-21],或者以此为基础进行一定的调整,间接地推动了干酪根分子模型的建立。
曹旭辉(2014)建立了分子式分别为C128H68O7,C406H528O19和C415H698O22的3种干酪根模型,用于不同类型干酪根对甲烷吸附机理的研究[20]。干酪根除了C,H和O元素以外,还含有诸如S,N,P等杂元素。大多数干酪根模型局限于C,H和O元素,并且在官能团,特别是支配原油裂解的长链往往也较为单调,离地质学家所需的干酪根模型有一定差距,难以贴近实际应用。刘向君等人(2017)建立了四川盆地下志留统龙马溪组干酪根的“平均分子结构”模型,其分子式为C206H158O19N4S4,实验结构参数和模拟的分子模型结构参数接近[60]。这些干酪根模型都引入了S和N这两种杂元素,为油气生成提供了很好的模型基础。然而众所周知,干酪根是生成石油和天然气的沉积有机质,是一种分散于沉积岩中的不溶性大分子,岩石中除了干酪根等有机组分外,还有无机矿物质,诸如石英、白云岩、方解石和伊利石等矿物组成以及一些微量元素Fe,Cu,Ni,Zn等等。因此,未来干酪根模型不能局限于有机组分,应当合理引入无机元素等,进而通过建立接近地下真实的沉积有机质模型,来探索油气成因机理。
分析前人建立的干酪根分子模型,一个普遍的特点是侧链链长较短,长侧链数量较少。无论是从实验还是实际的烃源岩抽提物和原油分析结果看,长链的正构烷烃占了相当的比例,常规的气相色谱分析(310 ℃)可以检测到37个碳原子的正构烷烃,高温气相色谱甚至可以检测碳数达73的高分子量正构烷烃。
由于经典分子动力学模拟在描述分子体系时,分子中原子之间的成键关系和电荷保持不变,无法直接用于生烃机理的研究。为了描述模拟体系中的化学反应行为,通常使用反应力场的势函数来描述模拟体系的化学反应过程,即反应分子动力学模拟。在众多的反应分子动力学模拟中,ReaxFF动力学模拟方法对于研究复杂的反应体系具有巨大的优势。ReaxFF力场的势函数基于键级,可以平滑地描述模拟反应体系中原子间的成键和断键过程,从而区别于经典的非反应力场(如COMPASS,Dreiding,pcff等),最重要的是不需要预先设定体系中的化学反应途径[61]。因此,ReaxFF动力学模拟方法是研究高温高压条件下复杂分子体系化学反应行为最具潜力的手段之一。
ReaxFF动力学模拟方法在燃烧和煤炭热解等领域中已被广泛应用,是一种非常可行的方法[61-63]。但在烃源岩干酪根生烃的分子模拟研究领域成果还非常少。Liu等(2015)采用ReaxFF反应力场,基于前人建立的Ⅰ型干酪根模型,对干酪根分子的热解生烃化学机理进行了初步研究,得出了许多有意义的认识:①页岩有机质的分解起始于C—O键的断裂;②自由基的稳定性决定了脂肪烃环开环的位置;③芳环上的长链取代基优先断链;④干酪根先裂解成C40+的高分子量烃,然后进一步分解成低分子量烃[64]。分子模拟的中间体和最终产物与实验室模拟产物基本吻合,表明了干酪根生烃的分子模拟方法是研究有机质生烃机理的有效方法。
未来干酪根生烃的分子模拟研究应从如下几方面入手:①采用化学分析与分子模拟相结合的方法,建立更可靠的、接近于真实的干酪根分子结构模型,比如在干酪根分子结构中加入Mg,Ni,V等金属元素;②采用实验模拟产物与分子模拟产物组成及性质对比的方法,选择合适的反应力场、选取合适的参数和算法;③建立物理模拟实验、分子模拟和地质实例相结合的综合研究方法,建立基于分子水平的微观尺度、实验室模拟的宏观尺度和地质尺度的联系,揭示烃源岩有机质的生烃机理、生烃特征及油气资源潜力。
6 原油裂解机理分子模拟
油藏中原油的裂解也是油气地球化学关注的重要基础研究领域。前人主要基于实验室模拟结果和化学反应动力学原理,对原油的裂解产物特征、化学反应过程及其控制因素进行了卓有成效的研究,取得了许多重要的认识。例如早期研究成果认为温度达到149 ℃,液态烃会全部裂解成气[56,65],而后来一些学者认为,在高温(160~200 ℃)条件下原油才会发生裂解[66-67]。Price等(1993)研究表明C15+重烃在Ro大于1.35%,甚至达7.0%尚可存在[66]。原油裂解和稳定存在的温度下限问题仍存在争议。
Waples等(2000)认为在实验条件下原油组成上的差异对原油裂解速率的影响较小[67],但在地质条件下,不同烃源岩类型的原油裂解生气的起始温度可能存在差别,例如Schenk等(1997)研究认为海相成因的原油比湖相原油开始裂解的温度要低大约10 ℃[68]。何坤等(2011)基于黄金管高温高压实验,对原油裂解研究表明,高压和水的存在能抑制原油裂解过程的自由基链反应,起到提高原油稳定性的效果[69]。粘土矿物尤其是蒙脱石或伊/蒙混层矿物,则会通过酸催化作用加速原油或烃类的裂解。帅燕华等(2012)通过原油、原油加水及各种水介质的恒温(350 ℃)热解模拟实验发现,地层水参与原油裂解成气过程,且水中的Mg2+对原油裂解反应有一定抑制作用[70]。
目前国内外对原油裂解过程的研究主要采用的是热模拟实验和化学动力学方法[71-72]。热模拟实验通常采用封闭黄金管高压釜体系,化学动力学参数模拟通常采用平行一级反应动力学模型,活化能服从离散分布,采取初次裂解模型或二次裂解模型进行模拟计算,应用较为广泛的是美国Lawrence Livermore国家实验室开发的Kinetics软件来拟合原油裂解实验产物的生成动力学参数。
尽管原油裂解实验模拟的方法已比较成熟,也取得较多的成果和认识,但目前对影响原油裂解的因素及其原理尚未弄清,比如原油组成的影响、岩石和矿物组成的影响、水在原油裂解过程中的作用,以及压力的影响等还有待于进一步探索。从实验研究结果看,部分认识不一致甚至存在相反的结论。而分子模拟作为一种有效的研究方法,可以从原子/分子水平上,认识原油裂解的微观机理,为解释原油裂解的化学原理、原油在油藏中的性质及保存、破坏机制提供证据,但目前为止,这方面的研究成果还很少。
原油裂解分子模拟的关键在于构建接近真实的原油分子模型和选择合适的反应力场。目前国内外学者对于原油分子模型的构建往往根据研究需求进行了相当程度的简化,仅仅是由单一或少量种类的原油中存在的小分子构成的简化模型。比如,Zhong等(2013)在研究原油中极性组分在水润湿二氧化硅表面的吸附行为时,分别以癸烷、甲苯、吡啶和乙酸作为原油模型[73]。Kunieda等(2010)在采用MD模拟研究芳烃通过弱氢键在轻质油-水界面发生自堆积时,构建的轻质油模型由8种烃类分子构成,分别是144个己烷、132个庚烷、156个辛烷、180个壬烷、96个环己烷、156个环庚烷、60个苯和156个甲苯分子,原油模型的组成和日本油田轻质油相关性较高,是一种典型的汽油模型[74]。可以看出上述原油结构模型对原油中化学组成的考虑并不全面,并不适用于研究原油裂解反应机理。
当前采用分子模拟方法研究复杂大分子的化学反应机理通常采用ReaxFF反应力场[61-63]。目前的成果主要针对原油中某些常见的单一化合物裂解机理的分子模拟研究[75-76]。例如Liu等(2018)采用反应分子动力学(RMD)和密度泛函理论(DFT)对烷基环己烷在高温下热解过程的初始和中间反应机理进行了研究。结果表明烷基环己烷初始热解主要有4种类型的反应路径:①烷基环己烷异构化形成C7H14双自由基;②甲基自由基的损失;③C—H键的断裂反应;④相邻C—C键的同时断裂[75]。Wang等(2010)采用反应分子动力学(ReaxFF MD)模拟和化学动力学模型研究了正十二烷的热解初始反应机制和动力学。研究表明正十二烷的热解初始机制主要通过2条途径:①C—C键的断裂来形成小分子的烃类自由基;②脱氢反应生成H自由基和相应的n-C12H25自由基。正十二烷热解初始反应生成的小分子自由基产物(包括·H,·CH3,·CH5)进一步发生去氢反应[76]。与基于正十二烷热解的一级动力学分析的实验结果相比,ReaxFF MD模拟可产生合理的Arrhenius参数,表明ReaxFF MD模拟可以对烃类燃料的热解初始机制和产物分布进行原子级的描述。
未来原油裂解机理的分子模拟研究应从如下几个方面考虑:①可先对单一原油组分进行有针对性的模拟,得到规律性认识,再由单一组分转向多组分模拟;②构建接近于真实、与所研究原油化学组成和性质基本相近的原油分子结构模型,可采用与干酪根分子模型构建类似的方法,构建非烃和沥青质的“平均分子模型”,根据原油饱和烃和芳烃组成实验分析结果,构建代表性的饱和烃和芳烃分子模型组合,最后根据原油的族组成,构建原油分子结构模型;③在建立真实、符合地质实际的原油分子模型基础上,可根据研究目的,在建模时适当突出刻画和放大原油中某些组分、某些特殊官能团或者化学键,可直观、深入和有针对性地研究原油裂解的化学机理;④介质和环境因素的影响,模拟不同矿物,包括粘土矿物、石英和碳酸钙等矿物作用的机理,以及压力的影响等;⑤分子模拟、实验室模拟以及地质实例解剖相结合,建立从微观尺度,到宏观尺度,再到地质尺度的联系和转换关系,从化学机理,到生烃动力学参数,再到油气资源类型和资源评价,将分子模拟结果用于油气勘探实践。
7 结论
1)油气地球化学分子模拟是当前有机地球化学涉及多学科交叉的前缘研究领域。煤层气和页岩气扩散机理、页岩油吸附分子模拟、油气运移分子模拟、干酪根生烃和原油裂解分子模拟是目前重点关注的方向。
2)计算机软硬件技术的发展,不同分子力场的开发以及大量地质地球化学资料的积累,为这一领域的发展进步提供了重要的支持。
3)分子模拟不仅为解决油气地球化学领域基础理论问题从原子、分子水平提供了微观机理方面的解释,也为非常规、深层-超深层等特殊复杂油气资源的勘探开发提供了新的理论支撑,具有非常广阔的应用前景。
猜你喜欢
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
建材发展导向(2021年14期)2021-08-23
航天工业管理(2020年9期)2020-12-28
航天工业管理(2020年1期)2020-04-20
中成药(2019年12期)2020-01-04
中国煤层气(2019年2期)2019-08-27
学苑创造·B版(2018年12期)2018-03-04
装甲兵工程学院学报(2017年4期)2017-09-16
火炸药学报(2014年3期)2014-03-20
