
UPLC-MS/MS同时测定动物源性食品中114种兽药残留
2021-08-15 13:54李晓芹丁洪流何新叶王伟
食品工业 2021年7期
李晓芹,丁洪流 ,何新叶,王伟
1.苏州市食品检验检测中心(苏州 215104);2.苏州市产品质量监督检验院(苏州 215104)
中国对畜禽产品、蛋奶制品等的需求日益增长,但违法用药造成动物源性食品兽药残留量超标,动物性食品安全状况不容乐观[1-2]。食以安为先,国家不断加大对食品安全的监管力度。但现有标准[3-6]多按照类别进行兽药残留检测,只能满足一类或少数几类兽药的检测,单一标准无法做到多种类药物覆盖[7],导致检测步骤繁琐、检验周期过长、检测成本偏高,从而降低监管效率。对动物源性食品中未知风险实施有效监控,需要建立尽可能全覆盖的兽药残留筛查方法[8]。
动物源性食品基质复杂,同时兽药残留量含量甚微、极性差别大。因此,样品前处理成为分析过程中耗时最长、产生误差最多的一个环节,是实现兽药残留高通量快速分析必须要突破的难点[9]。液质联用技术不断进步,因其高灵敏度、高选择性和抗干扰能力,推动兽药残留的分析从单一类别发展为跨种类高通量分析,同时降低对样品前处理的要求[10]。试验采用UPLC-MS/MS,通过一次提取净化,同时测定动物源性食品中磺胺类、喹诺酮类、大环内酯类、硝基咪唑类等11类114种兽药残留。该方法前处理技术简单,方法可靠,检测成本低,为实验室建立动物源性食品中兽药多残留检测技术提供经验和参考。
1 材料与方法
1.1 仪器与试剂
液质联用仪(I-Class Xevo TQ-XS,配电喷雾离子(ESI)源,美国Waters);高速冷冻离心机(Allegra X-30R,美国贝克曼);氮吹仪(N-EVAP型,美国Organomation);数显型涡旋混合仪(德国IKA);超纯水仪(美国密理博)。
乙腈(质谱级,德国Merck);甲醇(质谱级,德国Merck);甲酸(质谱级,日本TCI);试验用水为GB/T 6682规定的一级水;固相萃取柱(Oasis PRiME HLB,6 mL/200 mg,美国Waters);标准物质(纯度92.7%~100%,德国Dr.Ehrenstorfer或Witega)。
1.2 标准溶液的配制
分别准确称取10 mg(精确至0.01 mg)标准物质,用甲醇溶解定容,配制成1000 μg/mL标准储备液,棕色储液瓶存储,于-18 ℃保存,保存期1年。分别移取适量的标准储备液,配制成10 μg/mL的混合标准溶液,于-18 ℃保存,保存期6个月。
1.3 样品处理方法
1.3.1 对于鱼肉等易分散的样品
准确称取2.5 g(精确至0.01 g)均质后的试样置50 mL带盖塑料离心管中,加入10 mL 0.2%甲酸-水乙腈(20∶80,V/V)混合溶液,涡旋混匀提取1 min,超声提取2 min,按8000 r/min离心3 min后,取3 mL上清液上固相萃取柱,准确移取2 mL滤液氮吹至近干,用初始流动相定容至0.5 mL,超声溶解2 min,按10000 r/min离心5 min后取清液直接供质谱分析。需同时做空白试验。
1.3.2 对于猪肉、虾肉等不易分散的样品
准确称取2.5 g(精确至0.01 g)均质后的试样置50 mL带盖塑料离心管中,先加入2 mL 0.2%甲酸水溶液,涡旋分散后加入8 mL 0.2%甲酸乙腈溶液。按照1.3.1“涡旋混匀提取1 min……”的步骤继续提取净化。
1.3.3 对于牛奶、鸡蛋等含水量较大的样品
准确称取2 g(精确至0.01 g)摇匀后的试样置50 mL带盖塑料离心管中,直接加入8 mL 0.2%甲酸乙腈溶液,按照1.3.1“涡旋混匀提取1 min……”的步骤继续提取净化。
1.4 基质加标标准工作曲线
称取与试样基质相应的阴性样品(精确至0.01 g),加入适量的标准溶液,按照1.3的步骤和试样一同进行提取和净化。
1.5 色谱和质谱条件
1.5.1 液相条件
色谱柱,Waters Acquity UPLC BEH C182.1 mm×100 mm,1.7 μm;柱温45 ℃;进样体积2 μL;流速0.30 mL/min;流动相A为0.2%甲酸水溶液,流动相B为0.2%甲酸甲醇溶液。梯度洗脱条件:0 min 98% A,0.5 min 98% A,13 min 1% A,15 min 1% A,15.1 min 98% A,17 min 98% A。
1.5.2 质谱条件
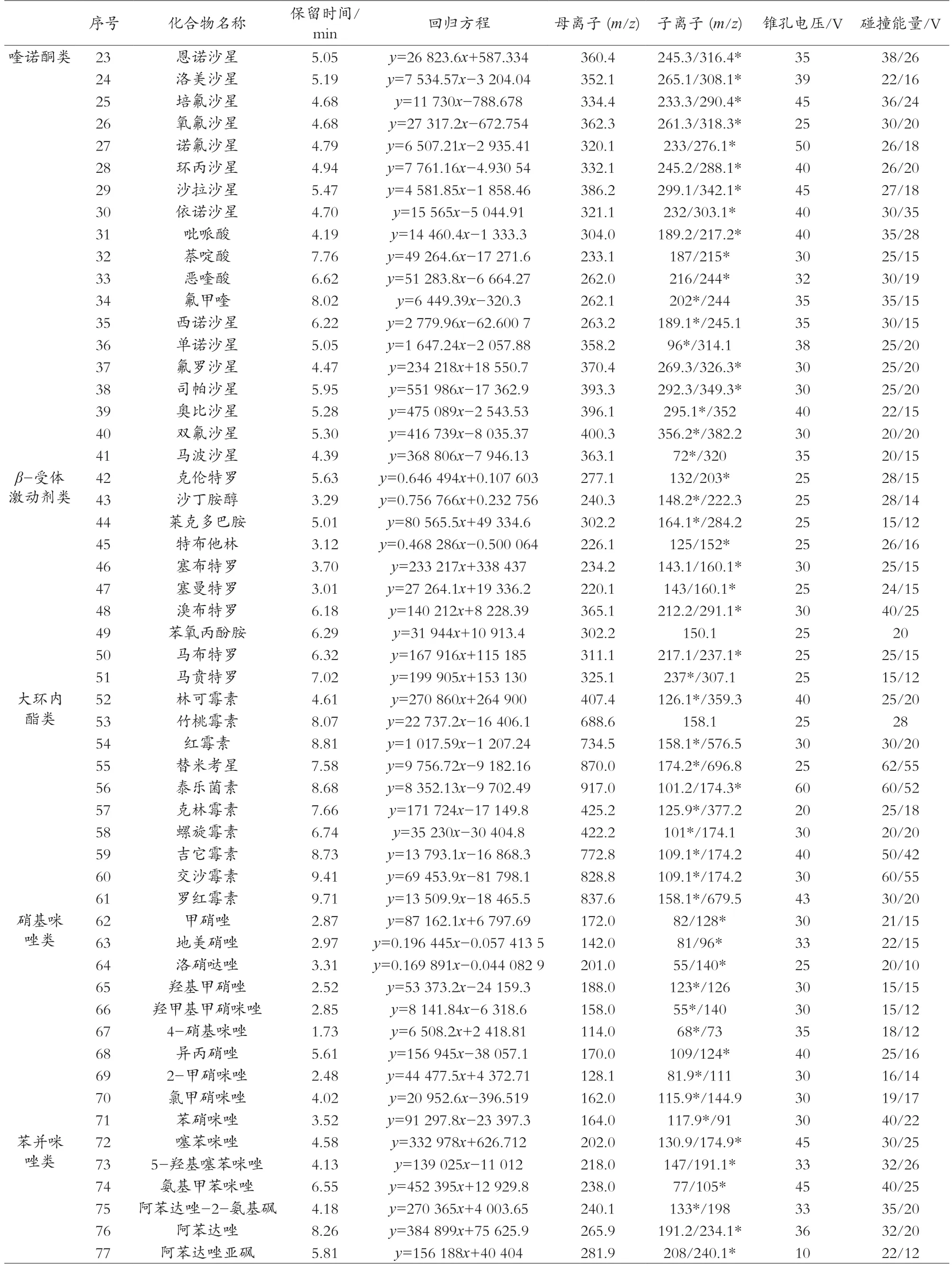
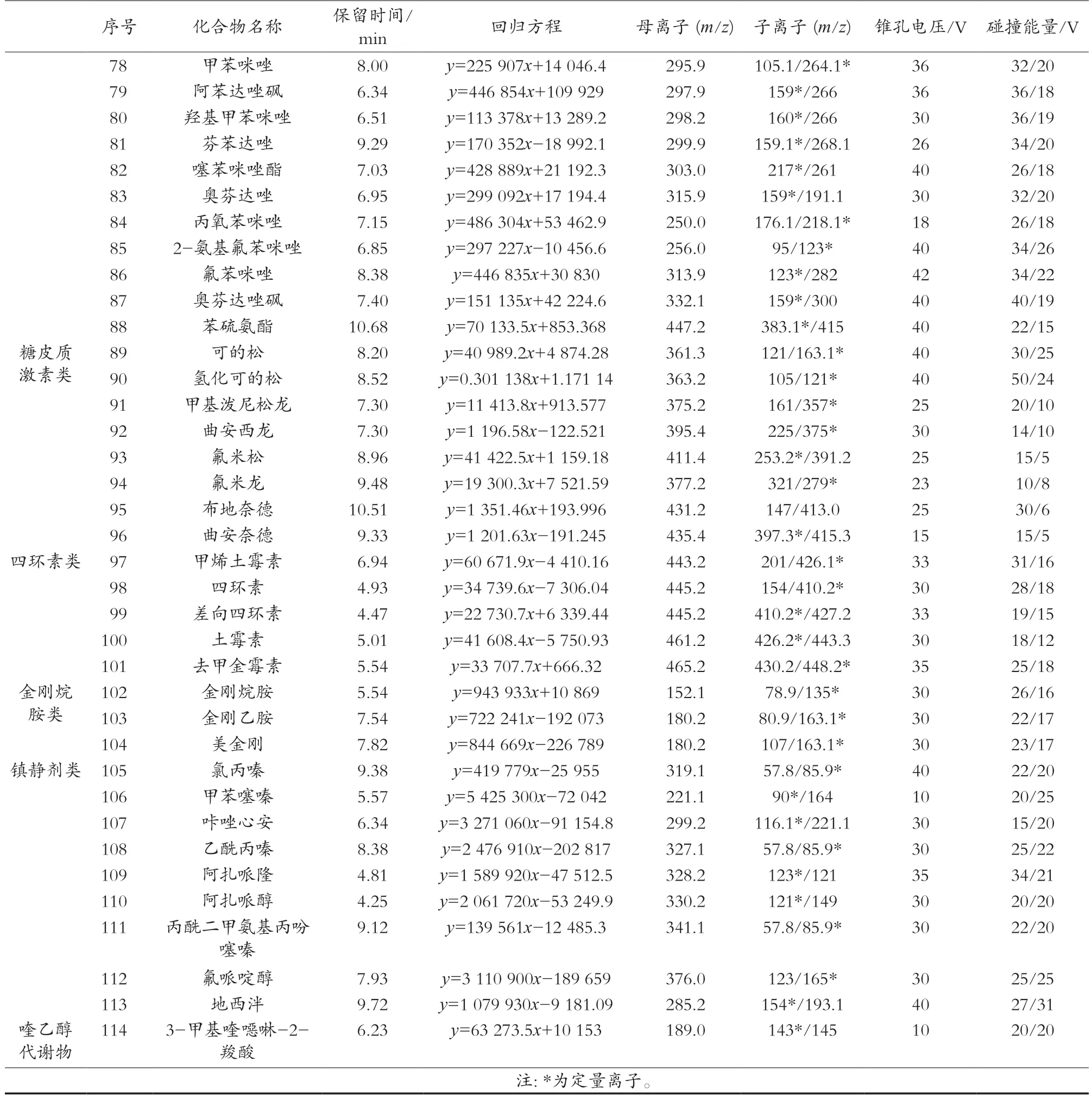
ESI+模式扫描,MRM采集模式;毛细管电压1.0 kV;离子源温度150 ℃;脱溶剂气温度600 ℃;脱溶剂气流速1000 L/h;锥孔气150 L/h;碰撞气流速0.15 mL/min。各化合物的离子对、锥孔电压及碰撞能量等质谱参数见表1。
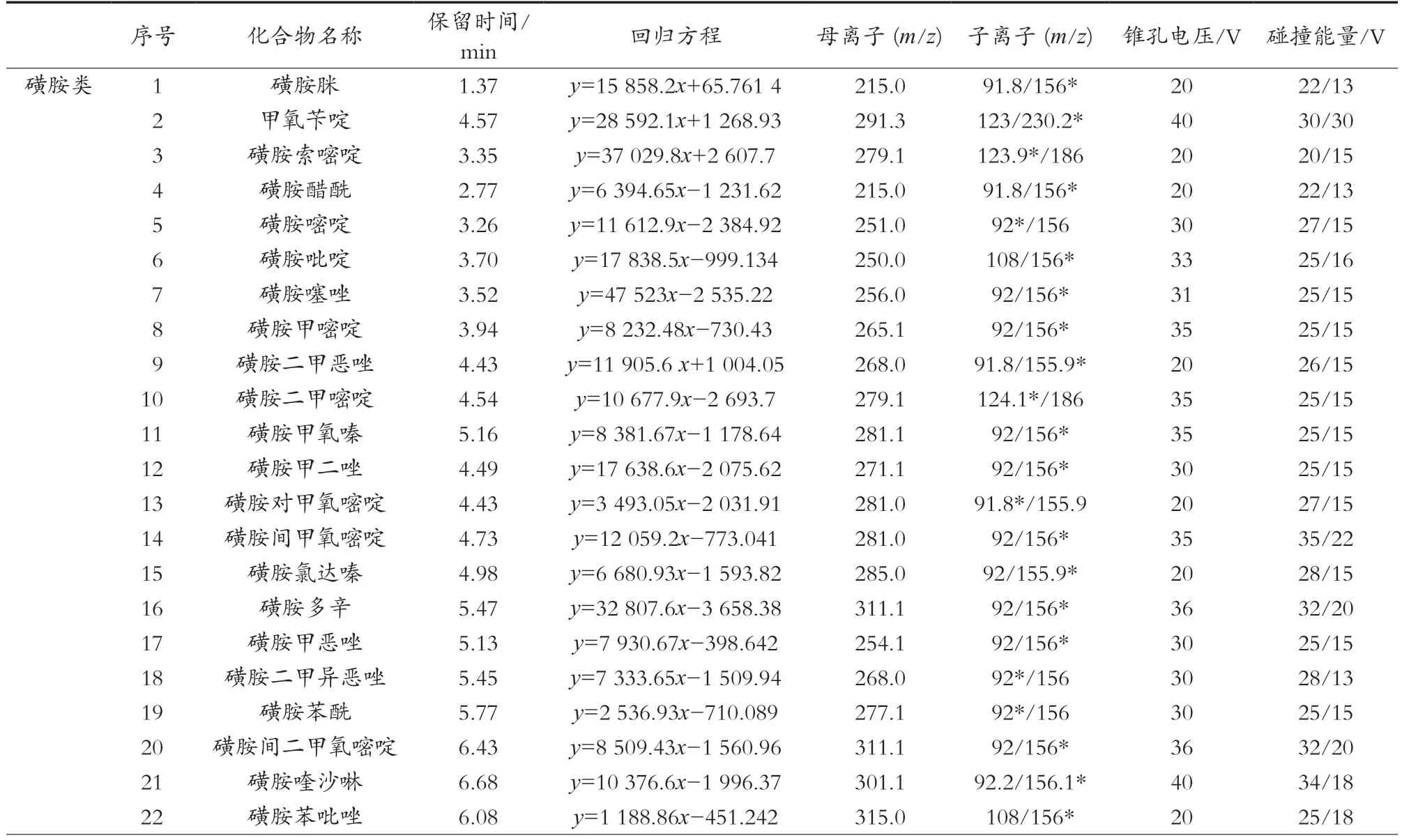
表1 114种化合物的MRM质谱采集参数及回归方程
2 结果与讨论
2.1 质谱参数优化
通过泵直接将标液注射入离子源,根据分子质量和带电方式,进行一级质谱分析,得到分子离子峰,确定各目标物的母离子,同时优化锥孔电压。对母离子进行二级质谱扫描,得到碎片离子信息,选取2~3个响应较高、稳定的特征性子离子,同时优化碰撞能量。
在进样模式下,连接色谱柱,通过优化毛细管电压、离子源温度、脱溶剂气温度、脱溶剂气流速、锥孔气流速等条件,选取合适的质谱条件。试验发现,毛细管电压对离子对丰度影响明显。比较毛细管电压在0.2~2.5 kV范围内目标物的响应强度,结果发现毛细管电压1.0 kV时,喹诺酮类、大环内酯类、β-受体激动剂和硝基咪唑类的标物响应强度整体较高,噪音低,磺胺类等其他化合物的响应强度可接受。
通过实际阴性样品基质配标和溶剂配标得到的图谱进行比较,选取干扰较小、信噪比最高的离子对进行监测,确定为定量和定性离子对。
多种化合物同时检测,采集通道增多,导致峰的采集点数变少,从而降低定量结果的准确度和重复性。根据待测物的保留时间(tR),进行分段采集(tR±0.5 min),以保证每个峰有足够的采集点数。
2.2 液相条件优化
2.2.1 流动相体系选择
考察不同种类的流动相体系,结果发现加入乙酸铵使峰形更加尖锐,但同时信号整体降低,喹诺酮类拖尾严重。加入0.2%甲酸使得喹诺酮类拖尾现象明显改善,可能是由于该酸性条件下喹诺酮类化合物溶解性更好。乙腈体系下色谱系统压力适中,但部分目标物峰形略差,有略微前伸或拖尾现象存在。甲醇有更强的洗脱能力,得到的峰形尖锐,响应强度略有提高,但系统压力比较高。因此,在满足信噪比的前提下,采用0.2%甲酸水-0.2%甲酸甲醇体系,降低流动相流速,以0.3 mL/min流速梯度洗脱,柱压适中,同时流动相更易配制,利于实验室提高检验效率。在此流动相条件下,负模式响应差,因此只选取正模式下响应强度满足要求的兽药。
2.2.2 流动相梯度优化
因同系物较多,例如磺胺甲氧嗪/磺胺间甲氧嘧啶/磺胺对甲氧嘧啶、磺胺二甲异恶唑/磺胺二甲恶唑、磺胺醋酰/磺胺脒、氟甲喹/恶喹酸、磺胺间二甲氧嘧啶/磺胺多辛、阿苯达唑砜/羟基甲苯咪唑、金刚乙胺/美金刚等,通过离子对不能完全区分,需优化流动相梯度,使其达到基线分离,利用保留时间进行定性。
MRM离子流图如图1所示,所有的目标物均在11 min之前出峰,1针进样的仪器分析时间17 min,缩短了检测时间。
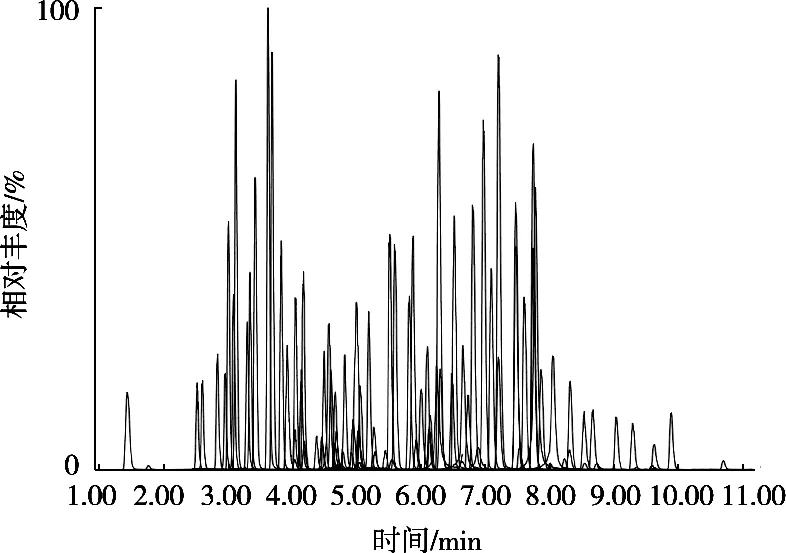
图1 114种药物MRM离子叠加图
2.3 样品前处理方法优化
样品前处理是兽药残留分析检测过程中耗时最长、产生误差最多的环节,直接决定整个分析检测方法的可行性和周期[11]。
涉及跨种类多残留检测时,液液萃取方法简单有效[12],缺点是基质干扰比较大,难以准确定量,且对色谱柱和仪器污染也较严重。分散固相萃取也经常被应用到兽药检测中[13-14],但步骤较多,需要多次离心和转移,试剂消耗量较大。试验通过比较,选取Oasis PRiME HLB固相萃取柱进行样品前处理,一步式净化操作简便,能够得到较好的回收率,满足日常分析要求。
乙腈是兽药残留检测中最常用的提取溶剂,能够提取出大多数的药物,同时引入更少的杂质[15]。试验比较纯乙腈、乙腈水(80∶20和60∶40,V/V)溶液和不同的甲酸含量(0.1%,0.2%和1.0%)时的提取效果[16]。结果显示:纯乙腈不利于含水量少的样品分散,样品易成团;1%甲酸沉淀效果最好,但磺胺类药物回收率偏低,0.2%甲酸溶液作为提取溶剂时,5类药物整体回收率较高且比较稳定,该结果和相关研究基本一致[17]。对于不易分散的样品,如猪肉,建议先加入水溶液,因为水具有良好的渗透性,可以较好地使样品分散。而水相占比提高使提取液混入更多的杂质,不利于净化。因此选用0.2%甲酸-乙腈水(80∶20,V/V)混合溶液进行提取。超声有助于样品分散和目标物的提取。牛奶等液体样品含水量大,直接加入0.2%甲酸乙腈溶液即可。
2.4 基质效应
动物源性食品中大量的蛋白和脂肪等进入提取液,净化不完全,会影响化合物在离子源中的离子化,使化合物的响应出现增强或抑制,出现基质效应[18]。基质效应影响数据的准确性,因此需要评价目标物在不同基质中的基质效应[19]。
通过基质加标和溶剂配标进行比较,结果表明,采用试验方法进行处理后,依然存在基质效应,尤其对喹诺酮类和磺胺胍、磺胺醋酰等药物低浓度点影响较大。因此,最终采用基质匹配标准曲线进行定量,即标准工作液加入空白基质,和样品一同前处理[20]。同时辅助同位素内标、降低上样量等手段[15]降低基质效应对定量结果的影响。
2.5 方法学验证
2.5.1 线性关系和定量限
分别在猪肉和鱼肉的空白基质中,添加一定浓度的标准溶液,与空白试验比较,获得10倍以上信噪比,确定此添加浓度为试验方法的最低定量限[21]。
将5个水平的标准溶液加入空白基质,提取、净化后,得到系列混合基质标准工作液,上机测定。以各目标物的定量离子的响应值为纵坐标,以质量浓度为横坐标绘制标准曲线,得到线性回归方程。线性系数r2均大于0.99,各化合物在相应的浓度范围内线性关系良好。114种兽药的定量限、线性范围见表2。
2.5.2 回收率与精密度
仅通过基质配标手段获得的回收率,不同化合物回收率在40%~90%之间,可用作快速筛查,但定量会存在一定误差。试验方法通过标液加到阴性样品中,和待测样品一同进行前处理后得到的基质加标标准工作曲线将回收率匹配到合理范围内[9]。
在猪肉、鱼肉中分别添加定量限、2倍定量限(2 LOQ)和10倍定量限(10 LOQ)3个水平的混合标准溶液,每个添加水平进行6次平行测定。114种兽药的定量限(测定低限)为0.2~2 μg/kg,线性范围为0.05~100 μg/kg,回收率为65.4%~114.9%,相对标准偏差(SRSD,n=6)均为0.6%~12.9%,具有良好的重现性,具体见表2。
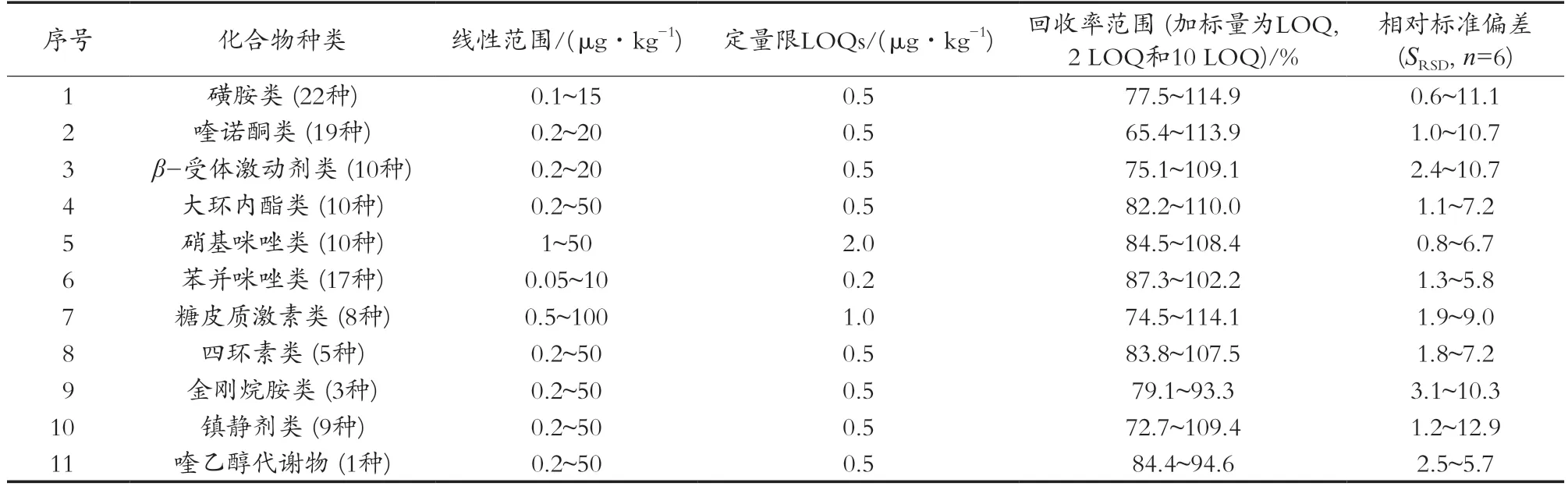
表2 11类药物残留的线性范围、定量限、回收率范围及相对标准偏差范围
2.5.3 实际样品的测定
实验室购买相应莱克多巴胺质控样品,使用试验方法进行处理和检测,最终结果在允许范围内。鱼肉糜中磺胺类药物残留能力验证获得满意结果。除猪肉、鱼肉样品,在鸡蛋、牛奶、蜂蜜样品中进行加标,均获得较稳定的回收率。对采用国标方法检测为阳性结果的样品,采用试验方法进行再次处理,结果基本一致。
3 结论
现有国家标准检测兽药残留,只能进行单一或一类兽药的分析,缺乏通用性和时效性,不能满足对兽药残留高通量分析的需求。试验建立同时测定动物源性食品中磺胺类、喹诺酮类、大环内酯类等11类114种常见兽药残留物的高通量筛查方法,减少繁琐的预处理步骤,仪器分析时间缩短至17 min,节省分析时间,提高检测效率,降低仪器成本。经验证,该方法能够用于实验室兽药残留的筛查工作,后续试验以期扩充药物种类,为实验室兽药残留检测技术进步提供参考。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
武警医学(2018年10期)2018-11-06
中国蜂业(2018年4期)2018-05-09
河南畜牧兽医(2016年24期)2016-11-29
当代化工研究(2016年6期)2016-03-20
湖南农业(2016年12期)2016-03-10
兽医导刊(2015年7期)2016-01-04
云南畜牧兽医(2014年4期)2014-02-28
无机化学学报(2014年3期)2014-02-28
无机化学学报(2014年3期)2014-02-28
