
富锂正极材料的高价态锰源前驱体制备与表征
2021-07-29 08:34:58毕亚凡
电源技术 2021年7期
刘 鑫,周 飞,毕亚凡
(武汉工程大学化学与环境工程学院,湖北武汉 430073)
近年来,由于新能源汽车需求和产量的持续扩张,对锂电池的能量密度提出了更高的要求[1]。而正极材料对锂离子电池的性能起着决定性作用,在已有的正极材料中,富锂锰基正极材料xLi2MnO3·(1-x)LiMO2(M=Ni,Mn,Co 等)因其比容量高,成本较低和低毒而被认为是极具潜力的锂离子电池正极材料[2]。由于普遍采用共沉淀法来合成富锂锰基正极材料,因此首次充放电不可逆损失较大,结构不稳定,倍率性能较差以及电压衰减等问题,尤其是充放电过程中因其晶相的转变或多晶球体的破裂而导致的连续放电容量和电压衰减[3],制约了该材料的商业化应用。为了解决上述问题,提高富锂锰基正极材料的电化学性能,人们在表面包覆[1]、体相掺杂[3]及合成新晶型[4-5]等方面做了许多工作,取得了较大的进展。目前,制备富锂锰基正极材料的锰源主要采用低价锰盐,势必会带来环境污染及高成本等问题,而人们往往忽视对锰源的选择的重要作用,实际上,锰源形态对正极材料的电化学性能有十分重要的影响[6-8]。Tabuchi 等[8]研究了锰源对正极材料电化学性能的影响,认为高价态的KMnO4制备的样品较低价态MnCl2制备的样品具有更好的电化学性能。Chang 等[9]利用控制结晶氧化法制备出高价态球形Ni1/3Co1/3Mn1/3OOH 前驱体,表现出比低价态的Ni1/3Co1/3Mn1/3(OH)2前驱体具有更高的活性。Li 等[6]以化学合成的α-MnO2为锰源并掺钾,制备了具有优良循环性能的富锂锰基层状正极材料。然而,化学法合成MnO2微粒的工艺过程控制条件要求高,废水产生量大,不易形成规模化生产,并且获得的MnO2晶粒粒径与活性不一,直接影响后续合成的正极材料的电化学性能。因此,为改进富锂锰基正极材料合成工艺及其电化学性能,开发一种实用的活性高价态锰源前驱体的制备方法十分必要。
本文选择市售电解二氧化锰作为原材料,采用焙烧-歧化法制备出高活性MnO2并以此作为前驱体与锰源制备富锂锰基正极材料,初步探求此类高价态锰源前驱体制备工艺及其对制备的正极材料的电化学性能的影响。
1 实验
1.1 实验材料与方法
实验材料:实验所用试剂LiNO3、Co(NO3)2·6 H2O 和H2SO4等均为分析纯,电解二氧化锰(EMD)为市售电池级。
焙烧:称取一定量的EMD 置于马弗炉中,分别设置为600、700 和800 ℃焙烧温度,在空气气氛下焙烧5 h,发生脱氧反应,得不同温度的焙烧样品Mn2O3。
歧化:分别将焙烧样品与质量分数20%H2SO4溶液以10∶1 液固比混合,置于烧杯经恒温磁力搅拌器70 ℃反应4 h,发生歧化反应,然后经过滤、水洗、干燥后得到歧化样品MnO2。含MnSO4滤液可循环套用,接近饱和后可作为生产电解二氧化锰的原料,无工艺废水产生。
正极材料制备:将LiNO3和Co(NO3)2·6 H2O 按照一定摩尔比配制成混合溶液,按设定的化学计量比称取一定量的上述歧化后的MnO2加入混合溶液中,置于恒温磁力搅拌器上搅拌1 h,恒温90 ℃。然后经120 ℃干燥4 h 后得混合固体样品,研磨均匀,再放入瓷烧舟,并置于箱式电阻炉中烧结,升温速率4 ℃/h,经500 ℃预烧4 h 后,再升温至750 ℃焙烧12 h,自然冷却至常温得富锂锰基正极材料。
1.2 电池组装
将富锂锰基正极材料、PVDF 和乙炔黑以质量比8∶1∶1 混合,加入一定量NMP 混匀成浆料,涂覆在铝箔上,在100 ℃真空烘箱中干燥12 h,制备成直径10 mm 的正极片。以锂片为负极,1 mol/L LiPF6/(DMC+EMC+EC,体积比为1∶1∶1)为电解液,Celgard2400 为隔膜,在高纯氩惰性气氛手套箱中组装成CR2032 纽扣电池。
1.3 材料表征和性能测试
材料物相分析采用德国Brucker 公司生产的D-8-Advance X 射线多晶体衍射仪。采集条件为Cu 靶,工作电压和工作电流分别为40 kV 和40 mA,扫描范围10°~80°;材料形貌表征采用日本电子株式会社JSM5510LV 型扫描电子显微镜(SEM)和德国蔡司GeminiSEM-300 型场发射扫描电子显微镜(FESEM);电池的电化学性能测试采用恒流充放电方式,Land 电池测试系统(武汉金诺电子有限公司生产)。
2 结果与讨论
2.1 XRD 表征与分析
2.1.1 焙烧前后的样品
图1 为EMD 以及经不同温度焙烧的EMD 样品XRD 谱图。
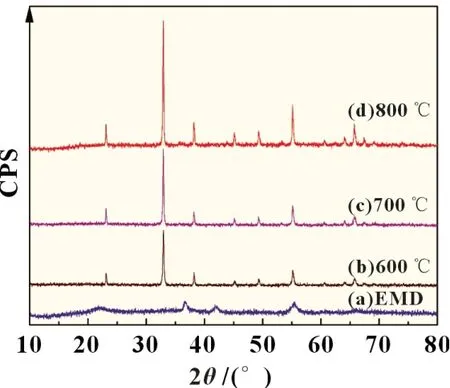
图1 不同条件热处理后EMD的XRD 谱图
由图1 中EMD 曲线(a)的衍射峰与标准卡片对比,在37.0°、42.0°和56.0°衍射峰与δ-MnO2的标准卡片(PDF#30-0820)一致,无明显杂峰,为纯度较高的δ-MnO2。600 ℃条件下,XRD 谱图出现了新的特征峰。曲线(b)在23.1°,32.9°,38.2°,49.3°和64.1°出现较明显的衍射特征峰,经比对与γ-Mn2O3标准卡片(PDF#41-1442)一致;在32.9°,45.2°,55.2°,65.8°和67.5°出现衍射特征峰,与α-Mn2O3标准卡片(PDF#73-1826)一致,且δ-MnO2特征峰消失,样品为纯度较高的Mn2O3。由图谱曲线(b)、(c)、(d)可知,随着热处理温度升高,生成的Mn2O3衍射峰变强,较为对称,这表示晶胞更为完整,晶体结构更有序,晶体结晶度高。因热处理温度升高使样品较易吸收能量完成晶型转变,利于晶体成核及成长,使Mn2O3结晶更为完整。而2 θ 在32.9°和38.2°处的衍射峰强度变强可能与γ-Mn2O3结晶更完善及新生成α-Mn2O3在该处的衍射峰与前者衍射峰的叠加效应所致,说明不同焙烧温度下所生成的Mn2O3中α 与γ 晶型含量不同。
2.1.2 歧化处理后的样品
图2 为不同温度下焙烧后的样品再经H2SO4歧化后的XRD 谱图。

图2 不同温度处理后的EMD歧化样品的XRD 谱图
由图2 中歧化样品曲线(e)的衍射峰与MnO2的标准卡片对比,在12.7°,18.1°,28.7°,37.6°,42.0°,49.9°,60.0°,69.6°呈现的衍射峰与α-MnO2标准卡片(PDF#72-1982)一致;在28.7°,37.6°,56.8°,72.5°呈现的衍射峰与β-MnO2标准卡片(PDF#72-1984)一致,而2 θ 在28.7°、37.6°处的衍射峰强度较强应是α-MnO2在该处的衍射峰与β-MnO2衍射峰的叠加效应所致,各歧化样品特征峰略有区别,样品为α 和β 的混合晶型MnO2。
根据XRD 谱图简单定量计算β-MnO2和α-MnO2的含量,参照PDF 卡片得到β-MnO2的参比强度(RIR)值为4.06,α-MnO2的RIR值为3.52。使用绝热法计算多晶型XRD 谱图中不同歧化样品中α-MnO2和β-MnO2混晶各自的含量。表1为不同温度下α-MnO2和β-MnO2混晶各自的含量。

表1 不同温度下α-MnO2 和β-MnO2 混晶各自的质量分数
由表1 可知,随着焙烧温度的提高,歧化样品中α-MnO2相对含量减少,β-MnO2相对含量增加。歧化样品(e)中α-MnO2相对含量为71.2%,歧化样品(f)中α-MnO2相对含量减少为66.6%,歧化样品(g)中α-MnO2相对含量下降为23.4%。推测原因:随着焙烧温度提高,δ-MnO2向Mn2O3转化所生成的α 与γ 晶型配比改变,从而引起歧化样品中α-MnO2和β-MnO2的比例不同。
2.1.3 合成的正极材料
将焙烧歧化处理后的不同锰源前驱体通过水热法将锂及过渡金属离子结合于MnO2模板中,经煅烧合成富锂锰基正极材料Li1.2Co0.26Mn0.54O2,其XRD 谱图见图3。

图3 各前驱体样品所制富锂锰基正极材料XRD谱图
由图3 可见,各样品20°~25°之外的特征峰均为α-NaFeO2层状结构,六方晶系,R-3m 空间群。谱图中20°~25°的微弱峰为超晶格特征峰,与过渡金属层及Li2MnO3内Li+排布的特征峰相同,对应空间群C2/m 的单斜晶胞结构[10],差异不明显。
2.2 SEM 表征与分析
2.2.1 焙烧后的样品
EMD 经不同温度焙烧后的SEM 表征图像见图4。

图4 不同温度焙烧后的电解二氧化锰的SEM图像
从图4 中可知,600 ℃焙烧样品(a)表面结构改变,形成很多细小裂纹;700 ℃焙烧样品(b)晶体颗粒表面裂纹明显增大;800 ℃焙烧样品(c)的Mn2O3晶体颗粒出现明显的三维网状结构,表明随着焙烧温度的升高,颗粒形成的裂缝也明显增大,焙烧过程发生的M-O 断裂的脱氧反应形成了Mn2O3,同时晶体发生重排,温度越高,提供的能量越大,重排程度越高,晶型结构也随之发生转化。
2.2.2 歧化处理后的样品
不同温度焙烧后样品再经H2SO4歧化处理后生成的二氧化锰SEM 表征见图5。

图5 不同温度处理的电解二氧化锰歧化后的SEM 图像
由图5 可知,经600 ℃焙烧后歧化所得MnO2材料为纳米级棒状颗粒,排布杂乱,长短尺寸不一,其比表面积巨大。随着焙烧温度升高,其歧化样品柱状颗粒尺寸不断增加。经800 ℃焙烧后歧化所制得的MnO2材料,柱形棒状晶体团聚明显,直径尺寸明显增大。说明EMD 晶体经焙烧脱氧后形成的Mn2O3经过硫酸歧化刻蚀,不稳定的Mn-O 键发生断裂与电子的转移,近一半的锰离子以二价离子形式进入溶液相,均能形成比表面积极大的MnO2。但是,原样品的焙烧温度不同,形成的晶粒形貌与晶型不同;而经酸歧化刻蚀后形成的晶粒形貌及晶型与对应的原样品直接相关,因此,不同焙烧温度处理的歧化样品应有不同的电化学活性。
由表面吉布斯公式可知,比表面积越大,其表面能越大。表面能的参与使纳米颗粒发生相变的外界热量减少,相变焓减少易于熔化。材料较大的比表面积利于熔融时锂盐及过渡金属离子向其内部渗透,这是促使活化样品具有高活性的重要原因。
2.2.3 合成的正极材料
由焙烧-歧化样品合成的富锂锰基正极材料FESEM 表征结果见图6。

图6 不同前驱体所合成富锂材料的FESEM图像
从图6 可知,制备正极材料均为单晶颗粒,有别于传统的共沉淀法所制备正极材料的多晶颗粒和二次球形颗粒结构,单晶颗粒机械强度较好,在充放循环过程中不易破碎,也改善了正极材料与电解质接触,有利于电荷转移。晶粒呈无规则排列,晶体棱角分明,结构完整。除未经处理的EMD 制备的样品外,其他样品粒径大约为100~200 nm,明显偏小,这可能与前驱体晶体粒径有关,与焙烧温度也相关。
2.3 电化学性能测试与分析
以不同前驱体合成的富锂正极材料Li1.2Co0.26Mn0.54O2组装成扣式电池,其充放电循环及倍率性能见图7 和图8。
从图7 中可知,未经处理的EMD 样品电化学活性最差,制备的正极材料放电比容量较低。600 ℃焙烧-歧化样品的电化学活性最好。随着焙烧温度的升高,所制备的正极材料电化学性能急剧降低,表明其歧化后的样品电化学活性也明显下降。800 ℃焙烧-歧化样品制备的材料首次放电比容量仅有123 mAh/g。而从图7 中可知,600 ℃焙烧-歧化样品制备的材料在前5 次的充放电循环过程中容量回升明显,放电比容量由227 mAh/g 升至279 mAh/g,放电比容量逐渐增加的原因可能与Li2MnO3层被激活有关,经过20 次循环后比容量为278 mAh/g,无明显衰减,其循环性能良好。此外,合成的各材料倍率性能见图8。显然,600 ℃焙烧-歧化样品在3C时的放电比容量仍可达139 mAh/g,也表现出较好的倍率性能。因此,600 ℃焙烧-歧化样品作为合成富锂锰基正极材料的锰源前驱体的化学活性高,且对其合成材料的电化学性能有明显的改善作用,值得深入研究。
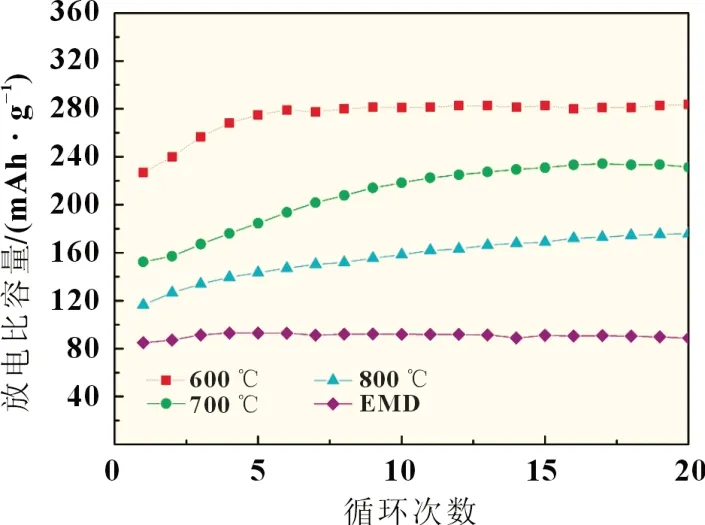
图7 各前驱体所合成富锂材料循环性能图
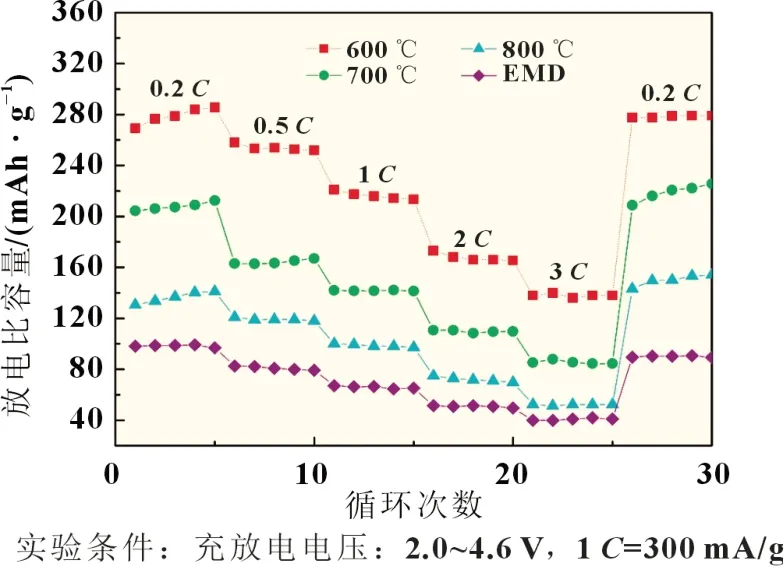
图8 合成的各富锂材料倍率性能曲线
3 结论
EMD 经不同温度焙烧后所得的Mn2O3晶粒经XRD 表征显示其晶型不同,随着焙烧温度的升高,生成的Mn2O3的X 射线衍射峰变窄变强,对称性变强,表明晶胞逐渐完整,晶体结构更为有序,扫描电镜显示其微观形貌也不同。此外,随着焙烧温度的提高,所制得焙烧样品经硫酸溶液歧化后,其样品中α-MnO2相对含量减少,而β-MnO2相对含量增加。经不同温度焙烧-歧化处理后均能制备出比表面积极大的MnO2晶粒,但是,经600 ℃焙烧再歧化样品α-MnO2含量最高,晶粒直径最小,为纳米级棒状晶粒。此外,以四种MnO2晶粒作锰源前驱体制备的富锂锰基正极材料的电化学性能测试表明,未经处理的电解二氧化锰样品化学活性最差,600 ℃焙烧再歧化样品化学活性最高,且对其合成材料的性能有改善作用,可作为制备富锂锰基正极材料的活性高价态锰源前驱体。
猜你喜欢
青岛科技大学学报(自然科学版)(2023年6期)2023-11-25 17:17:56
陶瓷学报(2020年2期)2020-10-27 02:16:14
汽车电器(2018年1期)2018-06-05 01:23:04
材料科学与工程学报(2016年1期)2017-01-15 13:33:52
当代化工研究(2016年7期)2016-03-20 16:21:54
电源技术(2016年2期)2016-02-27 09:04:42
中国塑料(2015年6期)2015-11-13 03:02:34
中国塑料(2015年8期)2015-10-14 01:10:48
电源技术(2015年12期)2015-08-21 08:58:54
电源技术(2015年9期)2015-06-05 09:36:06
