
CRISPR/Cas9基因编辑技术应用于绿僵菌
2021-07-26 10:44张二豪张杰
江苏农业科学 2021年11期
张二豪 张杰


摘要:近年来的研究显示,CRISPR/Cas9系统是强有力的基因编辑新技术。以蝗绿僵菌为试验对象,以同源重组敲除系统为对照,研究CRISPR/Cas9系统敲除蝗绿僵菌的基因isp4核苷酸序列。阐明了CRISPR/Cas9载体构建的方法,比较了CRISPR/Cas9和Recombinase敲除技术的异同,最后通过PCR和突变菌株的表型验证了CRISPR/Cas9系统能够应用于绿僵菌。结果表明,CRISPR/Cas9在昆虫病原真菌绿僵菌中是有效的基因编辑技术。
关键词:CRISPR/Cas9;重组酶;基因编辑;绿僵菌
中图分类号: S188 文献标志码: A
文章编号:1002-1302(2021)11-0048-06
收稿日期:2020-10-09
基金项目:周口师范学院高层次人才科研启动经费研究项目(编号:2018B180061);西藏自治区自然科学基金 (编号:XZ2018ZRG-19)。
作者简介:张二豪(1989—),男,河南平顶山人,硕士,讲师,主要研究方向为分子生物学。E-mail:1158496424@qq.com。
通信作者:张 杰,博士,讲师,主要研究方向为分子生物学与生物工程。E-mail:Zhangjiezk@qq.com。
分子遗传修饰和基因工程技术需要精确地改变基因组的核苷酸序列。随着基因修饰新技术的不断发展,某些基因可被敲除或被降低表达水平[1],这些技术为基因功能的研究提供了便捷的途径。基因编辑就是对细胞基因组中目的基因的核苷酸序列甚至是单个核苷酸进行替换、切除,增加或者是插入外源的DNA序列,使之产生可遗传的改变,进而研究其功能的手段[2]。绿僵菌的研究已有135年的历史[3],它作为一个昆虫病原真菌的模式丝状真菌,常常用来研究真核细胞的生物学过程、基因的相关表型和侵染昆虫致病的过程。
为了更好地研究病原真菌的基因特征,多种基因操作技術已经在绿僵菌研究中应用[4]。以位点为靶向的DNA内切酶是基因编辑技术中强有力的方法之一[5]。这些内切酶能在基因组范围内与靶向序列直接结合,进而使DNA双链断裂,断裂的DNA被修复过程中产生DNA序列的修饰。开发这些工具酶的起始方向集中在同源内切酶、锌指内切酶和转录激活内切酶类[6-7]。与传统突变体材料的获得方法相比,基因编辑技术能定向改变基因的组成和结构,具有高效、可控和定向操作的优点[8-9]。在目标基因DNA产生双链断裂[10]的基础上进行基因编辑是传统基因编辑技术的共同之处,但是各种基因编辑技术的原理及作用方式并不相同。
同源内切酶利用单一结构域识别和切割双链DNA[11]的基因编辑技术具有一定的局限性。CRISPR/Cas9是新一代的识别和绑定特异DNA序列为导向的基因编辑技术[12]。Generoso等首次报道,CRISPR/Cas作为特异的短间隔序列,成簇存在于大肠杆菌(Escherichia coli)基因组内的特殊短重复序列之中[13]。随后的研究表明,CRISPR位点在细菌和古细菌的基因组中分别占40%和90%,这些位点具有适应性的免疫功能[14]。在2012年,CRISPR/Cas9在链球菌(Streptococcus pyogenes)中,通过锚定crRNA 5′端的20 nt核苷酸序列行使功能[15]。经过优化的CRISPR/Cas9系统编码序列在哺乳动物细胞中可以高度激活[16-17]。在实际操作时,在sgRNA上5′ 端设计的20 nt核苷酸序列能够与目标基因完全互补,即可达到基因编辑的作用[18]。基因编辑技术CRISPR/Cas9在高等真核生物上已经成功应用[19-20]。CRISPR/Cas9介导的基因编辑过程实质上是非同源末端重组(non-homologous end joining,NHEJ)修复和同源末端重组修复(homology-directed repair,HDR)过程[21]。与传统基因编辑技术相比,CRISPR/Cas9系统最大的优点是易形成基因序列倍增,这些倍增区域在sgRNA的锚定下很容易突变[22]。如果2个sgRNA位于基因组侧的两翼,它们中间区域的基因组会被删除或发生颠倒[23-24]。通过双sgRNA可以进行基因组序列置换[25]。
CRISPR/Cas9系统的高效和便利,促使它很快地成为了细菌、植物和细胞培养时的基因编辑工具。CRISPR/Cas9可以直接导入到受精卵中进行早期的胚胎基因修饰,进而获得基因修饰动物[26-27]。在基因组编辑工具应用时,通常是将含有特殊启动子的CRISPR/Cas9质粒通过农杆菌介导法或电激法送到目的细胞内[27-28]。然而CRISPR/Cas9基因系统在昆虫病原真菌中的应用尚未见报道。
本研究以蝗绿僵菌为材料,同源重组敲除系统为对照,利用CRISPR/Cas9系统敲除蝗绿僵菌的基因isp4核苷酸序列。试验结果显示,CRISPR/Cas9敲除载体构建的方法和Recombinase敲除载体的构建技术不同,PCR和突变菌株的表型验证了CRISPR/Cas9基因编辑和Recombinase基因编辑技术一样成功应用于绿僵菌。
1 材料与方法
1.1 菌株和培养基
蝗绿僵菌(Metarhizium acridum)(CGMCC No.1877),将该菌株接种到1/4萨氏葡萄糖酵母培固体养基(1/4SDA)上,28 ℃,避光倒置培养。DNA提取时用液体培养基摇瓶培养,培养后经过真空抽滤获得菌丝体。
大肠杆菌(Escherichia coli DH5α,鼎国试剂公司),固体培养时,将转化的大肠杆菌菌株接种到LB固体培养基上倒置培养,培养基含有终浓度50~100 μg/mL的卡那霉素。绿僵菌转化时选择的培养基为细胞核分离基液(NIM)培养基,含有终浓度400 μg/mL的抗草丁膦(PPT)。选择性靶基因为PPT基因。
1.2 质粒
商业人工改造质粒CRISPR/Cas9 pGK1.1 (Puror)和同源敲除质粒PUC19,含有抗卡那霉素和PPT基因。
1.3 核酸操作
将含有抗性的大肠杆菌接种到液体LB培养基中,经过离心获得菌体,用质粒提取试剂盒(TIANGEN)获得质粒DNA。
将绿僵菌菌丝体用液氮研磨,然后用基因组DNA提取试剂盒(Omega)提取,Taq Plus DNA Polymerase (TIANGEN)进行PCR克隆,进而获得目标核苷酸序列。
目标片段和质粒的连接用NovoRecPCR一步定向克隆操作。其余的分子生物学技术按照参考文献[14]执行。
1.4 转化试验
根据转化说明书(Invitrogen)将改造的质粒转入到E.coli感受态细胞;用电转化的方法,将目标质粒转化到感受态根癌农杆菌中;然后根据参考文献将含有抗性质粒的脓杆菌和绿僵菌孢子接种于含有PPT的固体培养基上,进而获得转化的绿僵菌。
1.5 转化子纯化
转化后,被转化的目的菌株经过PCR验证,然后在筛选培养基上进行纯化培养,保存菌种以便后续试验。
2 结果与分析
2.1 CRISPR/Cas9和 Recombinase编辑系统的识别基础
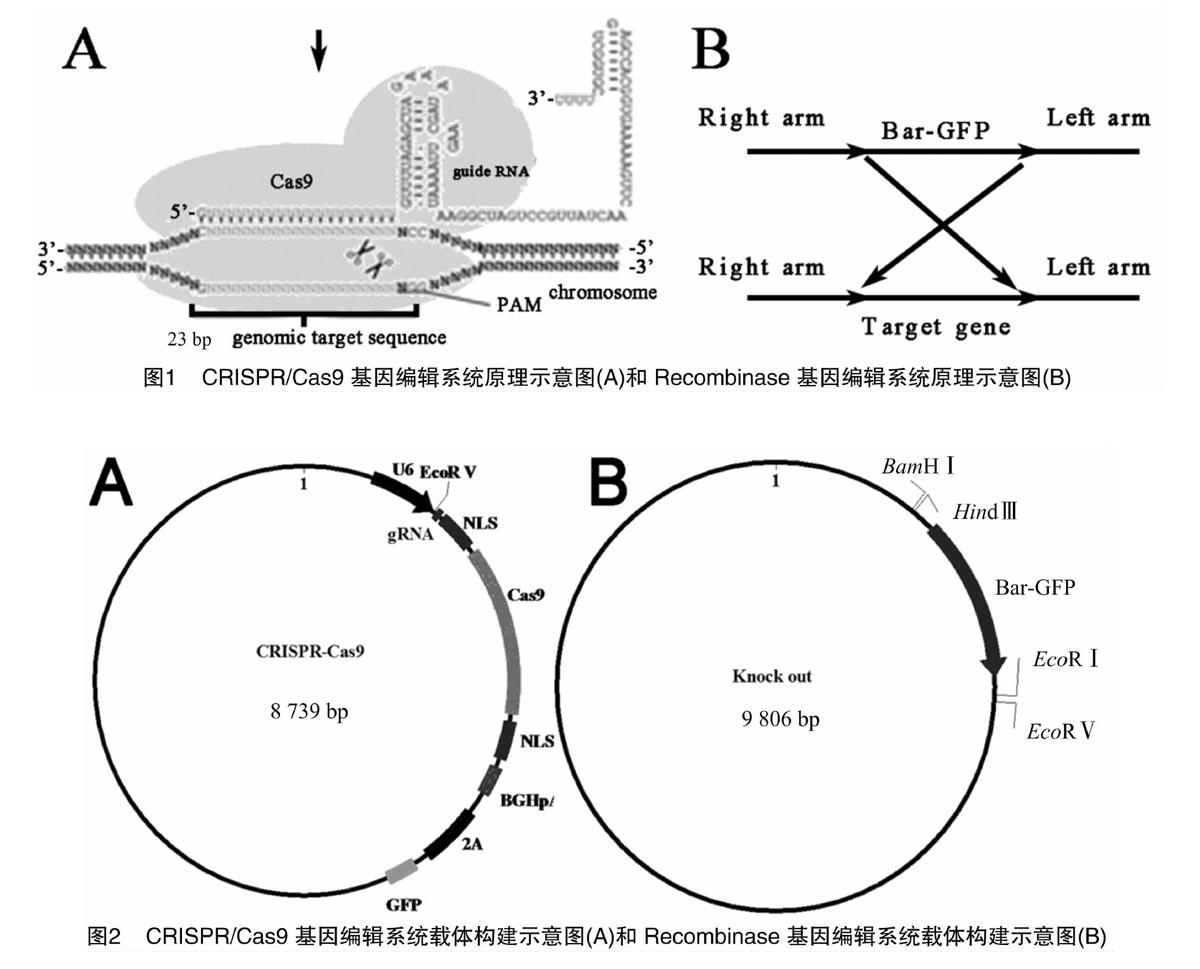
CRISPR/Cas9系统主要是依靠crRNA(CRISPR-derived RNA)通过碱基配对与tracrRNA(trans-activating RNA)结合形成双链RNA,此tracrRNA/crRNA二元复合体指导Cas9蛋白在crRNA引导序列靶定位点切断双链DNA。在基因组编辑过程中,tracrRNA和crRNA可以融合成为1条RNA(sgRNA)表达,同样可以起到靶向剪切的作用。靶向剪切的链缺口会促进同源重组,从而达到基因编辑的目的(图1-A)。对照Recombinase基因编辑技术主要依靠靶基因两侧左右臂引导生物本身的重组酶到靶位点进行基因编辑(图1-B)。由此可知,这2种基因编辑系统锚定目的基因位置的方式是不一致的。
2.2 CRISPR/Cas9和Recombinase基因编辑质粒的构建
基因编辑过程中,最重要的是构建一个高效的质粒载体。CRISPR/Cas9和Recombinase基因编辑系统的载体都是建立在商业质粒Puc19的基础之上。由图2-A可知,把优化的U6启动子(U6)、核定位基因序列(NLS)、Cas9蛋白基因、BGHpA、抗性基因和GFP序列共同整合到商业质粒pUC19中。在U6启动子后有一个靶基因酶切位点EcoRⅤ。Recombinase基因编辑系统是把抗性筛选PPT基因(含启动子)和GFP序列连接到商业质粒pUC19中(图2-B)。由此可知,CRISPR/Cas9基因编辑系统质粒的构建比Recombinase基因编辑系统复杂。CRISPR/Cas9基因编辑质粒含有多个基因敲除元件,而对照Recombinase基因编辑系统的质粒中只含有Bar基因序列和GFP序列。由此可知,构建的初始敲除质粒CRISPR/Cas9基因编辑系统比Recombinase基因编辑系统含有更多的复杂元件。
2.3 CRISPR/Cas9和 Recombinase基因编辑系统敲除isp4基因
为证明CRISPR/Cas9系统能够在绿僵菌上应用,经过生物信息学分析获得了绿僵菌性别分化基因isp4的mRNA序列(XM_007813236.1),作为敲除的目的基因。构建CRISPR/Cas9基因编辑载体时只需在该基因的mRNA编码序列中找到靶序列位点(GN20GG),然后在该序列的两侧加上接头 (F:TATATCTTGTGGAAAGGACGAT;接头R:GGTGCCACTTTTTCAAGTTGAT) 和gRNA序列即可。
通过CasFinder软件或word的查找功能均可找到以下19条靶序列(表1)。
以第1条靶序列为例合成如下的寡核苷酸序列:
casF:TATATCTTGTGGAAAGGACGATGGTATAGGCTTTTGCATCATCGGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCG;
casR:GGTGCCACTTTTTCAAGTTGATCGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAACCCGATGATGCAAAAGCCTATACC。
将上述引物交给公司合成PAGE 纯化寡核苷酸。然后寡核苷酸退火,退火后的寡核苷酸可以立刻使用或者在-20 ℃条件下长期保存。用EcoR Ⅴ酶切CRISPR/Cas9基因编辑载体致使其线性化,回收后即可根据NovoRecPCR一步定向克隆试剂盒说明书将上述退火后的寡核苷酸连接到线性化的CRISPR/Cas9 基因编辑载体上。阳性克隆用引物gRNA-F和gRNA-R进行 PCR验证时,能够扩增出180 bp的条带(图3-A),表明载体构建正确。
用生物信息学方法获得了绿僵菌性别分化基因isp4及其侧翼序列,该序列左臂含有EcoR Ⅴ (GATATC) 酶切位点和BamHⅠ(GGATCC)酶切位点,右臂含有HindⅢ (AAGCTT) 酶切位点。故构建Recombinase基因編辑载体时选用BamHⅠ和EcoRⅠ酶先后线性化载体。
根據NovoRecPCR一步定向克隆试剂盒说明书设计PCR扩增左右臂的引物和接头见表2。
经过PCR扩增获得敲除载体的左右臂,经过酶切后获得线性化载体,根据NovoRecPCR一步定向克隆试剂盒说明书构建成Recombinase基因编辑载体。经过转化的阳性克隆用引物isp4F和isp4R能够扩增出2 800 bp的DNA条带(图3-A)。
含CRISPR/Cas9和Recombinase载体的农杆菌侵染绿僵菌后获得阳性敲除绿僵菌孢子,荧光显微镜观察看到绿色荧光的绿僵菌孢子(图3-B和图3-C)。上述结果表明了isp4基因的CRISPR/Cas9和Recombinase敲除载体成功在绿僵菌中表达。
为一步证明绿僵菌敲除载体被正确敲除,用RT-PCR对isp4基因的表达量(引物RT-PCR-isp4F和 RT-PCR-isp4R)进行了验证(图4),菌落形态的观察也证明了2种敲除方法均改变了目的菌株的基因。这些结果表明isp4基因能够被正确敲除。
3 讨论
绿僵菌作为一种昆虫病原真菌的模式真菌,敲除技术限制影响了对其基因序列的大规模操作。CRISPR/Cas9基因编辑系统首先在细菌中被报道,随后被应用于动植物的基因靶向技术,不断得到优化[29-30]。CRISPR/Cas9系统的特异性被限制在sgRNA 5′端20 nt 的位点,这20 nt的核苷酸和靶向DNA进行Watson-Crick RNA-DNA容错性碱基配对,也有可能形成脱靶效应[22]。再者,多个物种的Cas9通过识别各自的sgRNA骨架,可在同一细胞中执行不同的功能,彼此之间互不干扰[9]。CRISPR/Cas9的特异性与靶序列的长度和构成及Cas9和sgRNA的浓度有关。sgRNA靶序列中鸟嘌呤和胞嘧啶含量过高或过低都会影响打靶效应[7]。
CRISPR/Cas9技术是基因编辑有力的工具,且被广泛应用,但它仍是相对新颖的技术,还有待提高。目前,CRISPR/Cas9技术的瓶颈是如何降低脱靶风险。正如上所述,降低CRISPR/Cas9的脱靶效应包括缩短sgRNA、用双切口dCas-FokI等方法。每条sgRNA序列都含有变化的脱靶位点,故在试验时,须要仔细分析影响脱靶的因素,降低脱靶效率。近年来积累了很多检测脱靶突变的方法,可以让研究者更有效地预测脱靶效应[10,26]。众所周知,基因编辑技术的效应与内切核酸酶在识别基因组上的特异位点相关。例如,转录因子及近年来研究的CRISPR/Cas9易结合于基因组DNA[21]。转录因子绑定和染色质重塑与组氨酸修饰有关[16]。由于基因组序列表观修饰和基因序列变化的复杂性,研究者仍然缺乏对CRISPR/Cas9的酶活性和DNA绑定对CRISPR/Cas9效果的认识[20,31]。
在本研究中,利用CRISPR/Cas9技术和Recombinase技术同时敲除了绿僵菌的isp4基因。通过比较发现,CRISPR/Cas9技术和Recombinase技术一样能够成功应用于昆虫病原真菌(绿僵菌)。在研究过程中发现,CRISPR/Cas9在绿僵菌中也存在效率不是很高的现象。CRISPR/Cas9技术在构建敲除质粒骨架时比Recombinase技术复杂,但是一旦建成熟骨架,后续进行大量基因敲除时就相对容易。对于CRISPR/Cas9的脱靶效应和转化效率方面尚需进一步研究。
参考文献:
[1]Blasco R B,Karaca E,Ambrogio C,et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology[J]. Cell Reports,2014,9(4):1219-1227.
[2]Canver M C,Bauer D E,Dass A,et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells[J]. Journal of Biological Chemistry,2014,289(31):21312-21324.
[3]Cho S W,Kim S,Kim J M,et al. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease[J]. Nat Biotechnol,2013,31(3):230-232.
[4]Choi P S,Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology[J]. Nature Communications,2014,5 (1):1-9.
[5]Cong L,Ran F A,Cox D,et al. Multiplex genome engineering using CRISPR/Cas systems[J]. Science,2013,339(6121):819-823.
[6]Deltcheva E,Chylinski K,Sharma C M,et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase Ⅲ[J]. Nature,2011,471(7340):602-607.
[7]Doench J G,Hartenian E,Graham D B,et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation[J]. Nature Biotechnology,2014,32(12):1262-1267.
[8]Doudna J A,Charpentier E. The new frontier of genome engineering with CRISPR-Cas9[J]. Science,2014,28(346):6123-6128.
[9]Esvelt K M,Mali P,Braff J L,et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing[J]. Nature Methods,2013,10(11):1116-1121.
[10]Frock R L,Hu J,Meyers R M,et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases[J]. Nat Biotechnol,2015,33(2):179-186.
[11]Fu Y,Sander J D,Reyon D,et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs[J]. Nat Biotechnol,2014,32(3):279-284.
[12]Fujii W,Kakuta S,Yoshioka S,et al. Zygote-mediated generation of genome-modified mice using Streptococcus thermophilus 1-derived CRISPR/Cas system[J]. Biochem Biophys Res Commun,2016,477(3):473-476.
[13]Generoso W C,Gottardi M,Oreb M,et al. Simplified CRISPR-Cas genome editing for Saccharomyces cerevisiae[J]. Journal of Microbiological Methods,2016,127:203-205.
[14]Hong M,Peng G,Keyhani N O,et al. Application of the entomogenous fungus,Metarhizium anisopliae,for leafroller (Cnaphalocrocis medinalis) control and its effect on rice phyllosphere microbial diversity[J]. Applied Microbiology and Biotechnology,2017,101(2):6793-6807.
[15]Horvath P,Barrangou R. CRISPR/Cas,the immune system of bacteria and archaea[J]. Science,2010,327(5962):167-170.
[16]Hwang W Y,Fu Y,Reyon D,et al. Efficient genome editing in zebrafish using a CRISPR-Cas system[J]. Nature Biotechnology,2013,31(3):227-229.
[17]Hynes A P,Lemay M L,Moineau S . Applications of CRISPR-Cas in its natural habitat[J]. Current Opinion in Chemical Biology,2016,34:30-36.
[18]Ishino Y,Shinagawa H,Makino K,et al. Nucleotide sequence of the iap gene,responsible for alkaline phosphatase isozyme conversion in Escherichia coli,and identification of the gene product[J]. J Bacteriol,1987,169(12):5429-5433.
[19]Kieler J B,Duong K L,Moye-Rowley W S,et al. Targeted gene deletion in Aspergillus fumigatus using microbial machinery and a recyclable marker[J]. J Microbiol Methods,2013,95(3):373-378.
[20]Kundaje A,Kyriazopoulou-Panagiotopoulou S,Libbrecht M,et al. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements[J]. Genome Research,2012,22(9):1735-1747.
[21]Kuscu C,Arslan S,Singh R,et al. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease[J]. Nat Biotechnol,2014,32:677-683.
[22]Lin Y,Cradick T J,Brown M T,et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences[J]. Nucleic Acids Research,2014,42(11):7473-7485.
[23]Luo S,He M,CaoY,et al. The tetraspanin gene MaPls1 contributes to virulence by affecting germination,appressorial function and enzymes for cuticle degradation in the entomopathogenic fungus,Metarhizium acridum[J]. Environ Microbiol,2013,15(11):2966-2979.
[24]Szewczyk E,Kasuga T,Fan Z . Efficient sequential repetitive gene deletions in Neurospora crassa employing a self-excising β-recombinase/six cassette[J]. Journal of Microbiological Methods,2013,92(3):236-243.
[25]Szewczyk E,Kasuga T,Fan Z . A new variant of self-excising β-recombinase/six cassette for repetitive gene deletion and homokaryon purification in Neurospora crassa[J]. Journal of Microbiological Methods,2014,100:17-23.
[26]Tsai S Q,Wyvekens N,Khayter C,et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing[J]. Nat Biotechnol,2014,32(6):569-576.
[27]Tulloch M. The genus Metarhizium[J]. Trans Br Mycol Soc,1976,66(3):407-411.
[28]Wan H,Feng C,Teng F,et al. One-step generation of p53 gene biallelic mutant Cynomolgus monkey via the CRISPR/Cas system[J]. Cell Research,2015,25(2):258-261.
[29]Wang H,Yang H,Shivalila CS,et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J]. Cell,2013,153(4):910-918.
[30]Wang J,Zhuang J,Iyer S,et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors[J]. Genome Res,2012,22(9):1798-1812.
[31]Weninger A,Hatzl A M,Schmid C,et al. Combinatorial optimization of CRISPR/Cas9 expression enables precision genome engineering in the methylotrophic yeast Pichia pastoris[J]. Journal of Biotechnology,2016,235:139-149.
