
锐钛矿型TiO2(101)面与Ag相互作用的第一性原理研究
2021-07-26 00:39:18胡洁琼普志辉恀彦亭
贵金属 2021年1期
沈 月,胡洁琼,普志辉,恀彦亭,潘 勇,闻 明
(1. 贵研铂业股份有限公司,昆明 650106;2. 西南石油大学,成都 610002)
TiO2因具有密度低、抗腐蚀性强、生物活性强、氧化还原能力强等诸多优点,成为了目前首选的舰船结构材料、生物医用材料及半导体光催化材料之一[1-3]。通过贵金属Ag的表面改性来调控TiO2的表面结构进而提高其性能是一种简单有效的方法。如Ag/TiO2梯度复合薄膜较未改性的TiO2耐磨性大幅提高[4];Ag改性的 TiO2具有良好的光降解性能和稳定性[5]。
目前,关于TiO2表面改性的理论计算也有大量报道。如Liu等[6-7]采用第一性原理研究了银团簇在金红石型TiO2表面吸附对体系电子结构的影响,发现借助带隙处界面态的表面等离激元共振可增强TiO2表面的光催化效果。Wang等[8]采用第一性原理计算,发现H2O2在锐钛矿型TiO2(101)表面解离为一种非常稳定的 OOH/H构型,该构型中的一个氧原子可以进入晶格并形成表面(O2)o从而改变光催化性能。
考虑到计算周期和目前实验中遇到的最常见的TiO2晶面,我们采用的 TiO2最强峰即锐钛矿(101)面和金红石的(110)面进行计算。前期本项目已采用第一性原理计算Ag与金红石型TiO2(110)晶面的相互作用[9]。本文将采用密度泛函理论的平面波赝势方法,进一步对锐钛矿型TiO2(101)面上键桥Ti和键桥O吸附Ag原子的物理参数进行计算,从理论上研究Ag/锐钛矿型TiO2复合材料中的Ag与锐钛矿型TiO2的相互作用。
1 计算方法
为了计算TiO2(101)面上两种键桥形式吸附Ag原子的电子结构,本研究采用第一性原理的密度泛函理论(Density functional theory,DFT)平面波赝势方法。根据密度泛函理论,单电子波函数的Schrödinger方程为:
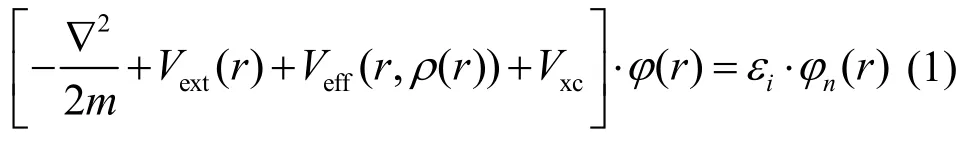
式中φn(r)是单电子波函数,Vext(r)为外场的势,Veff(r)为有效势:

包括体系中原子核对电子的库仑吸引势:

和电子之间的库仑势φ(ρ(r)),可以用电子态密度泛函数表示为:

Vxc为电子间交换相关势,体系的电子态密度泛函为:

其中an为第n个本征态的电子态占据数。
本研究中电子之间的交换关联势选取为局域密度泛函势(LDA),通过CA-PZ函数进行交换相关势的修正;TiO2(101)面吸附Ag原子的结构模型采用Pulay的密度混合方案进行几何优化,截断能取为360 eV,K点取6×6×4,SCF误差为 2.0×10-6eV。
2 结果与讨论
2.1 吸附能
因锐钛矿型TiO2(101)面上存在两种键桥形式,即键桥Ti和键桥O,Ag原子在不同的键桥形式上吸附能不同,而吸附能的差异又直接影响到Ag的吸附位置及稳定性。本文首先从吸附能大小来判断锐钛矿型TiO2(101)面上吸附Ag原子的稳定性,其吸附能的计算公式如下:
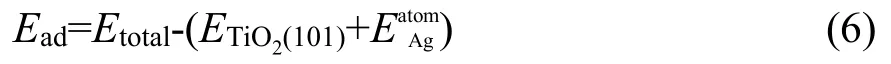
其中,Etotal为锐钛矿型 TiO2(101)面上吸附Ag原子的总晶胞能,ETiO2(101)的晶胞能,而为单个Ag原子的基态能。
Ag在锐钛矿型TiO2(101)不同键桥方式的吸附能,如表1所示。键桥O吸附Ag原子的吸附能为-1.375 eV,键桥Ti吸附Ag原子的吸附能为-1.619 eV。由此可知,键桥O吸附Ag原子的吸附能要大于键桥Ti的吸附能,换言之,当锐钛矿型TiO2(101)面上吸附Ag原子时,更容易以键桥Ti方式与Ag原子结合。

表1 Ag在锐钛矿TiO2(101)不同键桥方式的吸附能Tab.1 Adsorption energies of Ag in anatase TiO2(101) in different bond bridge modes
2.2 电子占据数、电荷布居和单位键长
表2~7列出了锐钛矿型TiO2(101)、键桥O吸附Ag原子以及键桥Ti吸附Ag原子模型各原子轨道上的电子占据数、电荷布居和单位键长。

表2 纯锐钛矿型TiO2(101)上各原子轨道上的电子占据数Tab.2 Electron occupancy of atomic orbitals on pure anatase TiO2(101)
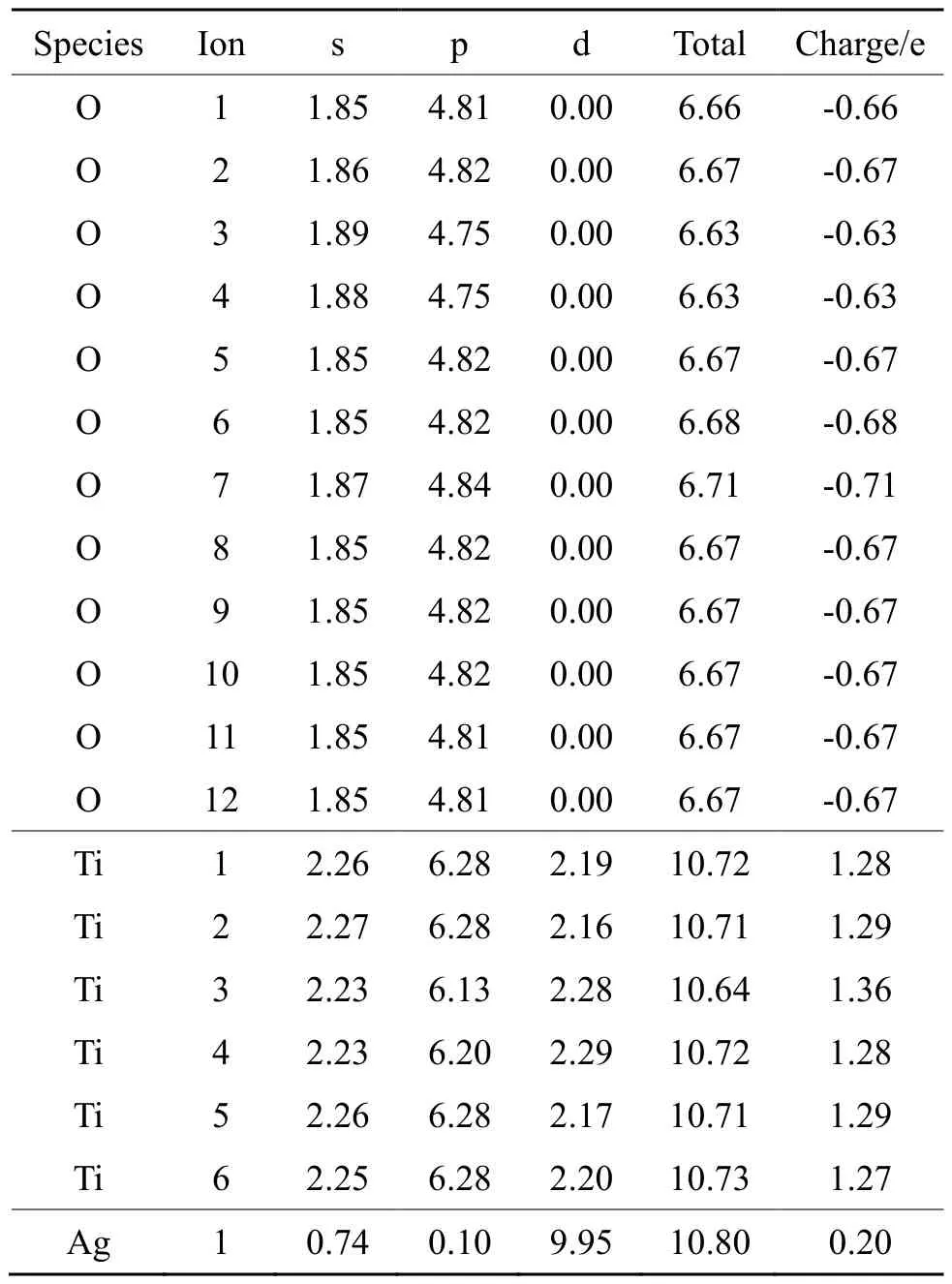
表3 键桥O上各原子轨道上的电子占据数Tab.3 Electron occupancy of atomic orbitals on O-bridge
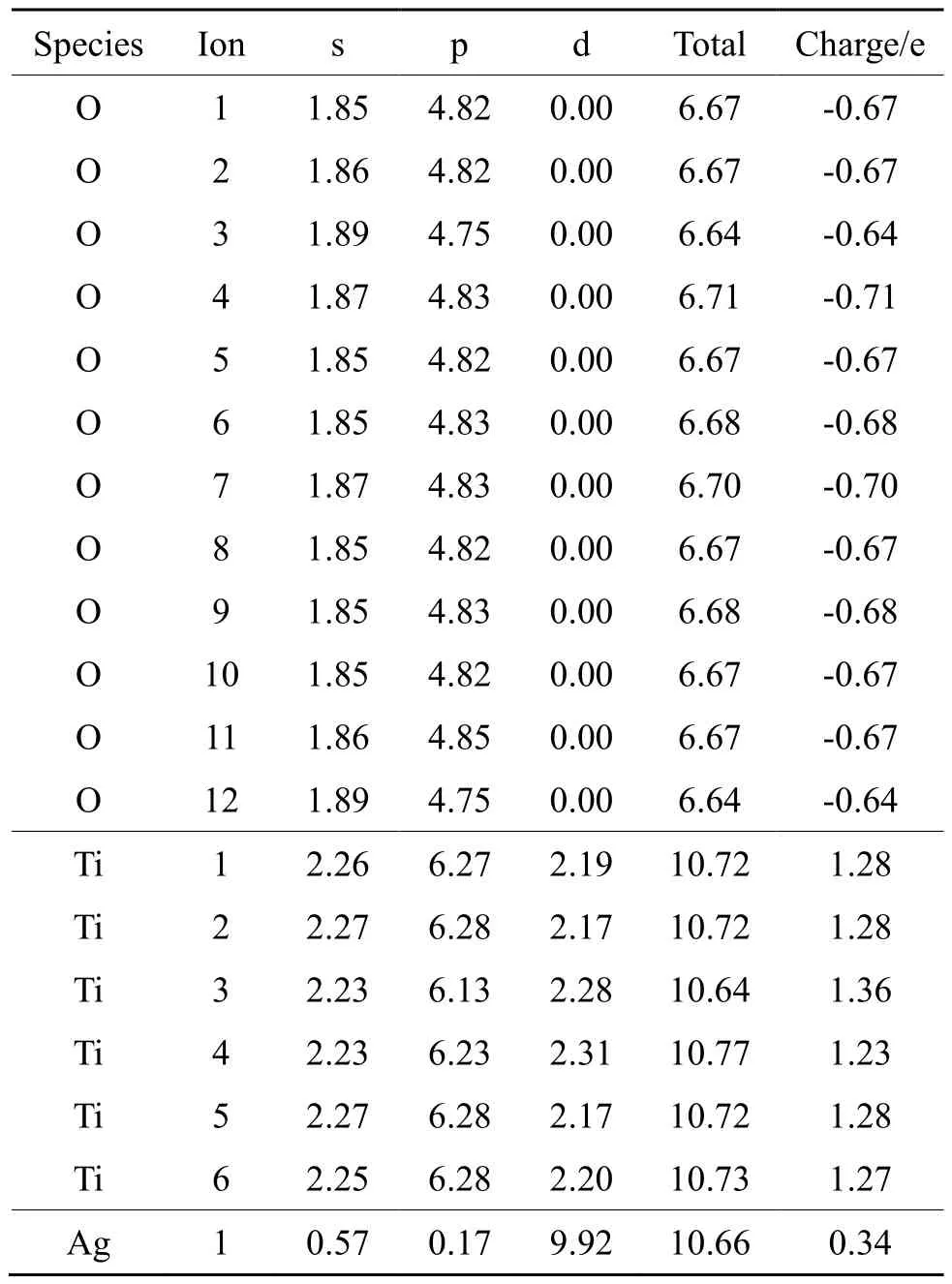
表4 键桥Ti上各原子轨道上的电子占据数Tab.4 Electron occupancy of atomic orbitals on Ti-bridge

表6 键桥O形式的电荷布居和单位键长Tab.6 Charge population and unit bond length on O-bridge

表7 键桥Ti形式的电荷布居和单位键长Tab.7 Charge population and unit bond length on Ti-bridge
锐钛矿型TiO2(101)、键桥O吸附Ag原子以及键桥Ti吸附Ag原子模型各原子轨道上的电子占据数如表2、3和4所示。各个原子轨道电荷布居发生了变化,即氧原子得到电子,而钛原子则失去电子;当锐钛矿型TiO2(101)面吸附 Ag原子时,Ag原子也是失去电子。从整体而言,外层轨道电荷总数基本保持不变,这遵守了电子守恒准则,但可看出各原子外层轨道上得失电子数是不一样的,这表明外层轨道上电子发生了spd杂化。值得注意的是,无论是键桥O形式还是键桥Ti形式的吸附,在吸附过程中Ag原子均是失去电子。这也意味着当锐钛矿型TiO2(101)吸附银原子时,将是O-Ag之间的电子转换。
由表5、6和7可知,纯锐钛矿型TiO2(101)面的Ti-O之间的电荷布居在0.36~0.46之间,而Ti-O之间的单位键长则为0.1805~0.1845 nm之间。当吸附银原子时,键桥O原子的O-Ag之间的电荷布居为0.11,而单位键长为0.2245 nm;而键桥Ti原子的 O-Ag之间的电荷布居为 0.11,但单位键长为0.2231 nm。两种键桥形式的电荷布居一致,但是键桥Ti原子的O-Ag之间的单位键长略小于键桥O原子的O-Ag之间的单位键长。从理论上而言,键长越短则原子之间的结合能力就越强,因此,键桥Ti上更容易吸附银原子,这与键桥Ti吸附银原子的吸附能分析是一致。
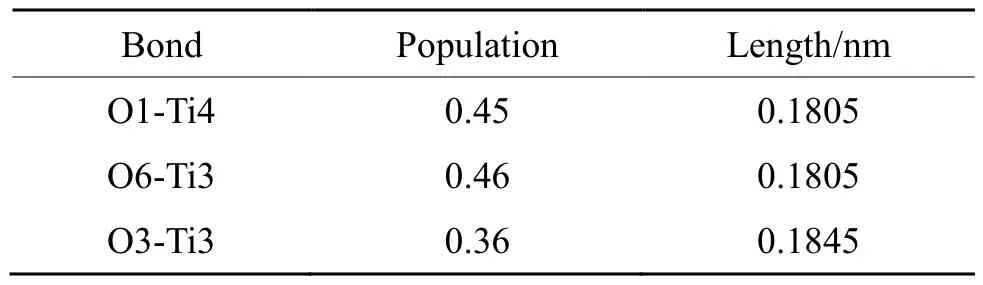
表5 纯锐钛矿型TiO2(101)的电荷布居和单位键长Tab.5 Charge population and unit bond length of pure anatase TiO2(101)
2.3 吸附Ag原子的电子结构计算
本节将从电子结构层次上分析锐钛矿型TiO2(101)面上的吸附形式来深入理解它吸附Ag原子的吸附性质。众所周知,原子之间相互作用的电子转移主要发生在各原子的价电子上,本文涉及的元素价电子分别为:O 2s22p2,Ti 3p63d24s2,Ag 4p64d105s1。图1为锐钛矿型TiO2(101)、键桥Ti形式和键桥O形式吸附Ag的总态密度分布图;图2为锐钛矿型TiO2(101)各原子的分波态密度图;图3分别为键桥Ti形式和键桥O形式吸附Ag的各原子的分波态密度图,其中图中横坐标零点处为 Fermi能级。
由图1~3可知,锐钛矿型TiO2(101)的态密度分布图主要是由O的2p电子轨道上的电子和Ti的3d电子轨道上的电子贡献。当锐钛矿型TiO2(101)吸附Ag原子时,则有Ag原子的4d轨道上的电子和少部分5s轨道上的电子参与贡献。对比图2和图3,发现在费米面附近键桥O形式的Ag原子的主峰峰值为11.32 eV,而键桥Ti形式的Ag原子的主峰峰值为7.75 eV,这说明键桥Ti形式的Ag原子失去的电子将多于键桥O形式的Ag原子,键桥Ti形式的 O-Ag之间将反应更为剧烈,即当锐钛矿型TiO2(101)面吸附Ag原子时,Ag原子更容易与锐钛矿型TiO2(101)面上键桥Ti上的O原子发生反应生成相应的化合物。

图1 锐钛矿TiO2(101)、键桥O和键桥Ti模型吸附Ag的总态密度分布图Fig.1 Total wave density states for Ag adsorption by anatase TiO2(101), O-bridge and Ti-bridge

图2 锐钛矿TiO2(101)中各原子的分波态密度图Fig.2 Partial wave density states of each atom in anatase TiO2(101)
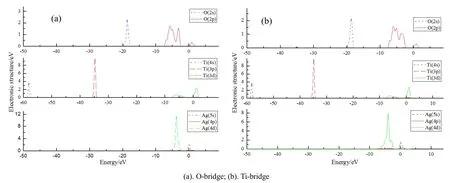
图3 键桥O和键桥Ti原子吸附Ag原子模型各原子的分波态密度图Fig.3 Partial wave density states of each atom on O-bridge and Ti-bridge
3 结论
第一性原理计算得到锐钛矿型TiO2(101)上键桥Ti和键桥O吸附Ag原子的相关物理参数表明:
1) 锐钛矿型 TiO2(101)吸附 Ag原子时,将是O-Ag之间的电子转换。
2) Ag原子更容易与锐钛矿型 TiO2(101)面键桥Ti上的O原子发生反应生成相应的化合物。
猜你喜欢
吉林大学学报(理学版)(2025年1期)2025-02-06 00:00:00
原子与分子物理学报(2021年1期)2021-03-29 07:29:40
原子与分子物理学报(2021年1期)2021-03-29 07:28:18
矿产勘查(2020年8期)2020-12-25 02:46:36
原子与分子物理学报(2020年5期)2020-03-17 07:00:14
无机盐工业(2017年6期)2017-03-11 14:43:47
材料科学与工程学报(2016年1期)2017-01-15 13:34:08
贵州师范学院学报(2016年3期)2016-12-01 03:53:53
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
