
PH-responsive strontium nanoparticles for targeted gene therapy against mammary carcinoma cells
2021-07-21 04:09:44AthirahBakhtiarEzharulHoqueChowdhury
Athirah Bakhtiar,Ezharul Hoque Chowdhury
a School of Pharmacy,Monash University Malaysia,Selangor 47500,Malaysia
b Jeffrey Cheah School of Medicine and Health Sciences,Monash University Malaysia,Selangor 47500,Malaysia
Keywords:Strontium Gene therapy Breast cancer SiRNA Nanoparticles
ABSTRACT Genetic intervention via the delivery of functional genes such as plasmid DNA (pDNA) and short-interfering RNA (siRNA) offers a great way to treat many single or multiple genetic defects effectively,including mammary carcinoma.Delivery of naked therapeutic genes or siRNAs is,however,short-lived due to biological clearance by scavenging nucleases and circulating monocytes.Low cellular internalization of negatively-charged nucleic acids further causes low transfection or silencing activity.Development of safe and effectual gene vectors is therefore undeniably crucial to the success of nucleic acid delivery.Inorganic nanoparticles have attracted considerable attention in the recent years due to their high loading capacity and encapsulation activity.Here we introduce strontium salt-based nanoparticles,namely,strontium sulfate,strontium sulfite and strontium fluoride as new inorganic nanocarriers.Generated strontium salt particles were found to be nanosized with high affinity towards negatively-charged pDNA and siRNA.Degradation of the particles was seen with a drop in pH,suggesting their capacity to respond to pH change and undergo dissolution at endosomal pH to release the genetic materials.While the particles are relatively nontoxic towards the cells,siRNA-loaded SrF2 and SrSO3 particles exerted superior transgene expression and knockdown activity of MAPK and AKT,leading to inhibition of their phosphorylation to a distinctive extent in both MCF-7 and 4T1 cells.Strontium salt nanoparticles have thus emerged as a promising tool for applications in cancer gene therapy.
1.Introduction
Extensive research on the genetic basis of human diseases with complete sequencing of the human genome has revealed many vital genes as possible targets in gene therapy programs,including cancer treatment.Cancer is the result of an accumulation of single or multiple gene defects,from down-regulation of tumor suppressor genes to up-regulation of proto-oncogenes and anti-apoptotic genes,promoting uncontrollable cell replication.Tumor suppressor genes are divided into two groups: promoters and caretakers.p53,an example of promoter tumor suppressors,is involved in inhibition of cell proliferation.It initiates transcription of Cdk inhibitor p21 and GADD45 that blocks cell cycle progress by acting as a general inhibitor of Cdk/cyclin complexes.Apoptosis of cells is facilitated through activation of many genes including BAX and NOXA,destabilizing mitochondrial membrane to assist cytochrome C release,triggering the apoptotic cascade of caspase activation.It is thought that more than 50% of mutated or missing p53 genes occurs in patients with cancer [1,2].Caretaker genes,including BRCA1 and BRCA2,ensure the integrity of the genome,especially in the DNA repair [3].MLH1 and MSH2 are another caretake genes that are involved in the mismatch of the DNA bases during the DNA repair.The mutation of the caretaker genes is associated with a dramatic increase in the rate of point mutation or microsatellite instability due to inability to remove the mis-paired nucleotides resulted from replication errors [4].The up-regulation of proto-oncogenes and antiapoptotic genes is associated with greater cell replication whereby oncogenes (mutated and cancer-causing forms of proto-oncogenes) typically increase the activity of encoded protein,driving the activity of cell growth or loss in the regulatory process to initiate proliferation genes such as MAP kinase and Ras [5].The up-regulation of anti-apoptotic genes(including Bcl-2 and Fas) enhance the survivability of the cells overexpressing the genes,therefore increases the opportunity to acquire the cellular mutation that eventually manifest into cancer [6].
Gene therapy is designed to modify cancer cells at the molecular level,in which many gene therapy strategies are being assessed through replacement of down-regulated genes associated with missing/non-functional gene activity,or down-regulation of the over-expressed genes via wild type tumor suppressor genes and small interfering RNAs(siRNAs) [7].Down-regulation of proto-oncogenes and antiapoptotic genes is induced by gene silencing activity to inhibit expression of specific genes that are involved in cell growth and proliferation,via the incorporation of endogenous siRNAs.siRNA modulates RNA interference process (RNAi) by causing nucleolytic degradation of the targeted mRNA,a post-transcriptional gene regulatory mechanism [8].
Successful delivery of genetic materials in the biological system may be impeded by many hurdles that exist in the blood circulation,which involves extracellular barriers such as endo-and exonuclease attacks,resulting in the short half-life of pDNA/siRNA in the circulatory system.The halflife of nucleic acid ranges from several minutes to an hour for siRNA and up to 21 min with pDNA upon parenteral delivery of naked genetic loads [9,10].Non-specific plasma protein interactions within the blood circulation causes premature degradation of genes by DNases and RNases that are abundant in the circulatory system.Reticuloendothelial system (RES) entrapment may also cause naked genes to be phagocytosed by mononuclear phagocyte system (e.g. Kupffer cells and macrophages) following intravenous delivery [11].Blood component interactions cause greater phagocytosed gene accumulation in RES organs,liver,and kidney,where the genetic materials were subjected to elimination.Negativelycharged nucleic acids electrostatically repel the anionic cell membrane,causing ineffective cellular uptake [12].
Intensive research in the last three decades has led to the development of many gene carriers,which are classified into two distinctive groups: viral and non-viral vectors.Viral systems are by far the most efficient means of genetic material delivery to mammalian cells.Extensively-studied viruses for gene delivery include retrovirus,adenovirus,adeno-associated virus and lentivirus [13].The successful application of viral particles is associated with their highly evolved and specialized structures comprising a protein coat surrounding a nucleic acid core.Highly organized viral structure enables genetic content to escape unwanted interactions with serum components,escape from endosomal cavity to be released from the viral load either before or after entering the nucleus [14].However,their marked immunogenicity causes the activation of the inflammatory response,leading to degeneration of transduced tissues.Production of viral toxin,insertional mutagenesis and limited DNA carrying capacity decapacitate their ability to effectively be the ideal carrier for nucleic acids.Additional production and packaging problem,along with high recombinant cost,further limit their successful applications in laboratory and clinical research [15].
Development of a non-viral approach devouring the beneficial virus-like properties and lacking the disadvantageous aspects has emerged as the most attractive approach to the delivery of gene materials.Non-viral vectors are generated from various biocompatible materials,which utilize innovative fabrication approaches to safely deliver the gene cargo [16].Negatively-charged pDNA/siRNA molecules should be ideally condensed with cationic reagents of nonviral structures to allow the formation of the complexes.The resulting composite interacts electrostatically with anionic heparan sulfate proteoglycans (syndecans) on the cell surface to reach the cytoplasmic side in the form of endosomes through the process called endocytosis[17,18].The most extensively studied non-viral carriers are liposomes,polymers and inorganic particles with potential for commercialization based upon clinical studies.Liposomes consist of a lipid structure with membrane-like surface,poses high affinity for cell membrane but has a short half-life in the circulatory system,associated with non-specific interaction with serum proteins.Polymers,including polyethyleneimine and poly(lactide-co-glycolide),are nontoxic carriers with great loading capacity with the downside of ineffective biodegradability and low transfection efficiency [19,20].Synthetic inorganic particles vary in size,shape and porosity,and possess the ability to protect the entrapped payloads from premature degradation.Inorganic particles,such as silica,gold and iron oxide are conveniently prepared by relatively simple chemical methods with flexibility to modify their surface with biological ligands to further improve their target specificity [21,22].However,many inorganic particles like gold and iron oxide are non-biodegradable,preventing their efficient post-endocytosis disintegration to release the loaded genetic materials [23].The generation of biodegradable nanoparticles with capability of efficient cytoplasmic release of payloads is therefore an important objective in ensuring high transgene expression and siRNA-mediated silencing activity.
Nanoparticles (NPs) have emerged as one of the novel inorganic gene delivery system for nucleic acid delivery approach.NPs are defined as solid carriers with the size of 1—1000 nm and has received considerable interest due to numerous advantages over many other carriers.It allows protection of the intended therapeutic cargo against premature degradation and helps to increase the accumulation to the targeted tissues by utilizing the unique characteristics of tumor microenvironment (TME).This approach will help in increasing the localization of genes that will increase the number of internalized genes for cellular interactions.NPs with the size of less than 500 nm can easily penetrate through the ‘leaky’ capillary system of malignant tissues due to a phenomenon known as ‘enhance permeability and retention (EPR) effect [19].Morphological changes in the lymphatic system of malignant tissues cause inadequate lymphatic drainage,resulting in greater retention time of the genetic content within the TME.NPs with sizes up to several hundred nanometers can enter the cells via membrane-bound vesicles,a process known as endocytosis.The process comprises of the following phases: formation of membrane vesicles with particle load,endosomal delivery of the therapeutic agents into the cell for distribution towards intracytoplasmic organelles [20].Precipitation reaction is one of the most recent approach in synthesizing nanoscale inorganic materials,which exhibit many unique and exciting physical and chemical properties with promising biodegradable activity.It involves the reaction of two soluble salts in an aqueous solution,forming an insoluble salt product [24].The desired chemicals react to produce a supersaturated solution,resulting in particle nucleation and ultimately into nano-sized particles [25,26].Carbonate apatite (COAP) and few inorganic barium salt NPs developed by this method demonstrated biodegradable activity with pH-sensing approach [27-29].The salt particles disintegrate effectively upon exposure to acidic environment of the endosome to release the therapeutic contents.Particle dissolution causes accumulation of cations and anions in the compartments,which might creating osmotic pressure across their membrane,resulting in swelling and rupture of the membrane to facilitate release of pDNAs or siRNAs into cytosol [30].Despite this,cytoplasmic activity of the payloads (pDNAs and siRNAs) upon disintegration of COAP and barium NPs remains relatively low,encouraging us to investigate different inorganic salts to possibly overcome this disadvantage.
Strontium (Sr) compound,an alkaline earth metal has recently been found to be beneficial for patients with osteoporosis and fractures,often act as calcium substitution for hydroxyapatite particles [31].Sr inhibits bone resorption while simultaneously stimulating bone growth,six times more efficient than placebo.Sr ranelate,a combination of Sr with ranelic acid,was found to be aiding bone growth,increasing bone density,and lessen vertebral,peripheral and hip fractures [32].Studies by Ravi et al.revealed that incorporation of Sr into bone cement in vivo,improved bone formation and decreased bone resorption [33].Sr is also used for superficial radiotherapy for bone cancer treatment due to beta emission and long half-life [34].Sr has been brough to the attention of many researchers owing to its their safety and efficacy profiles.A study done by Qian et al.demonstrated the effectiveness of Sr carbonate as the carrier for etoposide,where etoposide-Sr carbonate complexes displayed high loading ability and encapsulation efficiency in vitro [35].Additionally,the release of etoposide from its carrier was highest at pH 3.0,therefore potentially being pH responsive.The study of genetic vehicle is more challenging as compared to conventional small molecule drug delivery as the genetic materials are easily removed from the circulatory system.Application of inorganic Sr as part of the delivery structure for nucleic acids is therefore the main focus of this study,where their ability to efficiently adsorb pDNA and siRNA,transport the loaded genetic materials into mammary carcinoma cells,increase the effect on silencing of target protein expression following intracellular delivery and influence on cytotoxicity after delivery of MAPK siRNA and p53 gene were investigated through two different breast cancer models.
2.Materials and methods
2.1.Materials
Strontium chloride (SrCl),sodium sulfate (NaSO),sodium sulfite (NaSO),sodium fluoride (NaF),sodium bicarbonate(NaCO),sodium phosphate (NaHPO),4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES),ethidium bromide(EtBr) and propidium iodide (PI) were obtained from Sigma-Aldrich (Missouri,USA).Dulbecco’s Modified Eagle Medium(DMEM) was purchased from Gibco BRL (Tennessee,USA).Plasmid pGL3 (Promega,USA) containing luciferase gene under SV 40 promoter and plasmid pEGFP-N1 containing gene fluorescence protein (PGFP) gene were extracted from DH5 α bacteria strain of Escherichia coli (E.coli) using Qiagen plasmid extraction kit (Hilden,Germany).AF 488 siRNA(fluorescence siRNA) was also purchased from Qiagen (Hilden,Germany).
2.2.Cell culture and seeding
MCF-7 and 4T1 cells were cultured in 75 cmflasks in DMEMsupplemented with 10% fetal bovine serum (FBS) (Sigma Aldrich,USA),10% HEPES and 50μg/ml of penicillin at 37°C in a humidified 5% CO-containing atmosphere.Upon 90% confluence,sub-culturing was performed followed by trypsinization of the adherent cells and the detached cells were transferred in appropriate number in a new plate with 10% serum-supplemented DMEM.The cells from the exponential growth phase were seeded at 50 000 cells per well into a 24-well plate.
2.3.Fabrication and screening of inorganic strontium particles
Each inorganic Sr salt particles was prepared by incorporating 5μl of 1M cation-providing SrClsalt into 10μl HEPESbuffered solution (pH adjusted to 7.5),followed by mixing of the solution with 5μl of 1M of anion-providing salt,NaSO,NaSO,NaF,NaCOor NaHPO.The mixture was incubated for 30 min at 37°C and subsequently added to 10% FBSsupplemented DMEM medium to obtain the final volume of 1 ml particle solution.Absorbance at 320 nm wavelength was measured for all inorganic Sr salts and COAP particles (as control) spectrophotometrically (UV 1800 Spectrophotometer,Shimadzu,Japan) to observe the particle turbidity.Preparation of COAP NPs involves mixing of exogenous 5 mM CaClwith 1 ml bicarbonate-buffered DMEM (pH 7.4) and incubation for 30 min at 37°C,followed by addition of 10% FBS to the generated COAP particles to prevent extensive particle aggregation.The spectrophotometric measurement was conducted in triplicates and the data was plotted into a graph with mean ±SD.
Observation on the effect with changes in concentration of reacting salts to generate inorganic Sr particles was performed using similar turbidity measurement after mixing of SrClranging from 2 to 10μl of 1M into HEPES buffer solution with subsequent mixing of 5μl of 1M NaSO,NaSO,NaF,NaCOor NaHPOwith the resultant solution and incubation for 30 min at 37°C.Time—dependent analysis of particle formation was performed by changing the incubation time,varying from 0,30 to 60 min after adding 5μl of 1M SrClto 10μl HEPESbuffered media (pH 7.5) and mixing with 2μl of 1M NaSO,NaSO,NaF,NaCOor NaHPO,with temperature remains 37°C,followed by mixing of serum-supplemented DMEM to provide the necessary volume for observation and to slow the particle growth.The effect of pH on particle formulation was investigated by preparing HEPES-buffered solutions of various pHs,ranging from 4 to 9,before mixing with 5μl of 1M SrCland subsequently,2μl of 1M NaSO,NaSO,NaF,NaCOor NaHPOto incubate at 37°C for 30 min.Influence of incubation temperature was identified through cultivation for 30 min at selected temperatures (4°C,37°C and 70°C) of the generated salt particles following mixing of 5μl of 1M SrCland 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPOin HEPES buffered media at fixed pH (pH 7.5) for time of incubation (30 min).COAP were prepared as positive control in all experimental models.Studies were conducted in triplicates and analysis was shown as graphs with mean ±SD.
2.4.Size estimation and zeta potential measurement of Sr particles
Size and zeta potential measurement of fabricated Sr salt particles via ZetaSizer (Malvern,Nano ZS,UK) was obtained following mixing of 5μl 1M SrCland 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPOin a HEPES buffer,followed by incubation at 37°C for 30 min.The generated salt crystals were maintained on ice to prevent further particle growth prior to size measurement via ZetaSizer using a special capillary cuvette.A refractive index (RI) ratio of 1.325 (measured with DMEM media by refractometer) was used to measure the average particle sizes and zeta potential.Analysis of data was carried out using ZetaSizer software 6.20 and all samples were shown as mean ±SD.
2.5.Binding affinity of pDNA and siRNA towards Sr particles
Binding affinity of pDNA and siRNA towards various Sr salt particles was explored,involving qualitative and quantitative measurement of the fluorescence-labeled pDNA and siRNA complexed onto particle structures.1μg pDNA (pGFP) was labeled non-covalently with PI at 1:1 ratio before being added to 5μl of 1M SrCland subsequently to 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPO.After being topped up to 10μl with HEPES-buffered solution,the resultant mixture was incubated at 37°C for 30 min.DMEM medium was added to generate the final volume of 1 ml particle suspension.COAP particles were prepared as a positive control according to the same procedure as mentioned in methodology Section 2.3 .Microscopic visualization of the aggregated particles was observed following addition of the fabricated salt crystals to each well of a 24-well plate(Nunc,Denmark),where particle images were captured under fluorescence microscope (Olympus,Japan).Quantitative measurement of PI-labeled,pGFP-loaded Sr particles involved multi-label plate reading of the supernatant representing the unbound fraction of DNA (Victor X5,Perkin Elmer),following centrifugation of the differentially formulated particles at 15 000 RPM for 5 min.100μl supernatant was aspirated and transferred into 96-well plate (Nunc,Denmark),prior to fluorescence intensity examination.Binding affinity of siRNA for particles was determined via multi-label plate reading of the supernatant (denoting the unbound segment of siRNA),following incorporation of 100 nM AF 488 negative control siRNA (fluorescence siRNA,Qiagen,Germany) into 5μl of 1M SrCl,10μl HEPES-buffered solution and 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPO,prior to incubation at 37°C for 30 min.Each investigation was performed in triplicates and analyzed with mean ±SD.
2.6.Cellular uptake of pDNA and siRNA-loaded Sr particles
MCF-7 cells from exponentially growth phase were seeded at 50 000 cells per well into 24-well plates the day before transfection.1μg pDNA (pGFP) was labeled non-covalently with PI at 1:1 ratio and added into 5μl of 1M SrClin 10μl HEPES-buffered solution,followed by supplementation of 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPOinto the solution to form respective salt precipitates carrying fluorescent pGFP (incubation at 37°C for 30 min).10 nM AF 488 siRNA was introduced (Qiagen,Germany) to 5μl of 1M SrClfollowed by mixing with 10μl HEPES buffer and 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPO,and incubation at 37°C for 30 min to form 1 ml final volume of particle solution.10% FBS was finally added to the suspension to stop the particle growth.Sr salt complexes was subsequently incubated with seeded mammary carcinoma cells for 4 h,followed by removal of particle suspension,washing of the cells with 10 mM EDTA in PBS and addition of 100μl of serumsupplemented media.Fluorescence intensity was observed using a fluorescent microscope via FITC filter.
2.7.Visualization of Sr particles size and morphology via FE-SEM
The size and morphology of selected Sr samples were observed through Field Emission-Scanning Electron Microscope (Hitachi S-4700 FE-SEM,Japan).Sr particles were centrifuged at 15 000 RPM for 10 sec,followed by removal of supernatant and resuspension with milli-Q water.Each particle suspensions were maintained under ice to reduce particle growth prior to microscopic observation.1μl of each sample was placed onto carbon tape-coated sample holder and dried at room temperature,followed by platinum sputtering of the dried samples for 30 sec.Microscopic observation of the sputtered Sr particles was visualized at 10—15 kV.
2.8.Determination of cytotoxic activity of Sr particles
Cytotoxicity determination of selected Sr salt particles was performed via cell viability (MTT) assay,following incubation of treated cells for 24,48 and 72 h.5μl of 1M SrClwas added with 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPOin 10μl HEPES-buffered solution.Following 30 min of incubation at 37°C,MCF-7 and 4T1 cells were treated with the generated Sr salt complexes,with COAP NPs and untreated cells included as positive and negative control,respectively.The fraction of the viable cells was determined through MTT assay.Briefly,50μl of MTT (5 mg/ml in PBS) was added aseptically to the treated cells in each of the wells,followed by incubation at 37°C and 5% COfor 4 h.Medium containing MTT was aspirated after the incubation period and the formazan crystals formed at the bottom of each well were dissolved by addition of 300μl DMSO.Absorbance of the resulting formazan solution was determined spectrophotometrically at 595 nm wavelength using microplate reader (Dynex Opsys MR,USA) with reference to 630 nm.
2.9.Dissolution activity of Sr particles in acidic environment
1 M hydrochloric acid (HCl) was added to 1 ml of the Sr salt particles prepared through incubation of the respective salt reactants at 37°C for 30 min,to observe any changes in turbidity as a result of a gradual decline in pH from 7.5 to 6.5,5.5,4.5 and 3.5,mimicking the changes in pH within the endosomal cavity following endocytosis.The pH of the particle suspension was monitored upon addition of each drop of HCl until it reached the designated pH,where the amount of the particles was measured in terms of turbidity changes using a spectrophotometer.The result of every pH adjustment was obtained via absorbance at 320 nm wavelength.The salts were compared individually with COAP particles.The dissolution activity was performed in triplicates,expressed at mean ±SD.
2.10.Transgene expression activity associated with pDNA-loaded Sr particles
The suspension (1 ml) containing each Sr particles loaded with pGFP or luciferase reporter vector (pGL3)-carrying plasmid and supplemented with DMEM media was introduced into each well with approximately 50 000 MCF-7 or 4T1 cells seeded on the previous day.1μg pGFP or pGL3 was mixed with 5μl of 1M SrClbefore addition of 2μl 1M NaSO,NaSO,NaF,NaCOor NaHPOto generated selected Sr salts,SrSO,SrSOand SrFin 10μl HEPES solution through incubation at 37°C for 30 min.Serum-supplemented DMEM medium was added to achieve 1 ml of salt suspension.COAP was similarly prepared following addition of pDNA and 5 mM exogenous CaCl 2 into prepared 1 ml DMEM media,and incubation at 37°C for 30 min,prior to addition of 10% FBS to the suspension.The cells were treatedwith various fabricated formulations of Sr salt particles for a period of 4 h,before removal of the particle-containing media and brief washing of the treated cells in 10 mM EDTA in 1 ×PBS.Following substitution with 1 ml serum-containing DMEM media,the cells were further incubated for 48 h,prior to observation of gene expression through fluorescence microscopy (pGFP) and luciferase reported assay (pGL3) using a commercial kit (Promega,USA)and photon counting (Beckman Coulter,USA).Quantitative luciferase assay was repeated thrice and expressed in a graph as mean ± SD of luminescence activity/mg of protein.
2.11.Cytotoxicity assessment following intracellular delivery of p53 and MAPK siRNA using selected Sr particles
The suspension (1 ml) containing selected Sr particles loaded with p53 plasmid supplemented with DMEM media was introduced into each well with approximately 50,000 MCF-7 or 4T1 cells seeded on the previous day.1μg p53 plasmid was mixed with 5μl of 1M SrClbefore addition of 2μl 1M NaSO,NaSOor NaF to generate selected Sr salts,SrSO,SrSOand SrFin 10μl HEPES solution through incubation at 37°C for 30 min.Serum-supplemented DMEM medium was added to achieve 1 ml of salt suspension.COAP was prepared following addition of pDNA and 5 mM exogenous CaCl 2 into prepared 1 ml DMEM media,and incubation at 37°C for 30 min,prior to addition of 10% FBS to the suspension.The cells were transfected with various formulations for a period of 4 h,before removal of the particle-containing media and brief washing of the treated cells in 10 mM EDTA in 1X PBS.Upon substitution with 1 ml serum-containing DMEM media,the cells were further incubated for 48 h,The effect of p53 gene expression on cell viability was evaluated by adding 50μl of MTT (5 mg/ml in 1 × PBS) solution to the treated cells.After 4 h of incubation,medium containing MTT was aspirated,followed by dissolution of the formazan crystals in each well by mixing with 300μl DMSO solution.Medium containing naked siRNA and COAP represented both negative and positive control,respectively,for the study.Qualitative measurement of formazan crystals in the form of optical density (OD) was observed at 595 nm wavelength with reference to 630 nm via microplate spectrophotometer (Bio-Rad,USA).
The knockdown activity of an endogenous gene and expression/activation levels of the associated signaling molecules following intracellular delivery of a particular siRNA using selected Sr particles were demonstrated.10 nM MAPK siRNA was introduced into 5μl of 1M SrCl,followed by incorporation of 2μl of 1M NaSO,NaSOor NaF,in 10μl HEPES media forming SrSO,SrSOand SrFcomplexes,before incubation at 37°C for 30 min.Serumsupplemented DMEM media was used to top up the particle suspension to 1 ml.MAPK siRNA-loaded COAP (positive control) was prepared by addition of MAPK siRNA and 5 mM exogenous CaCl 2 to 1 ml of DMEM medium and incubation at 37°C for 30 min.10% FBS was subsequently added to the suspension.Both MCF-7 and 4T1 cells were treated for 4 h,followed by removal of medium,washing with EDTA in PBS and substitution with 1 ml serum-supplemented DMEM medium.Subsequent incubation was performed for 48 h,before addition of 50μl of MTT to the cells in each well to form formazan crystals by metabolically active cells.Medium containing MTT was aspirated post incubation,with formazan crystals in each well dissolved by mixing with 300μl DMSO.Medium containing naked siRNA (no salt) represented the negative control of the study.Quantitative measurement of formazan crystals in the form of OD was done at 595 nm wavelength with reference to 630 nm using microplate spectrophotometer (Bio-Rad,USA).The cell viability of siRNAloaded particles and naked siRNA was calculated based on equation:

Each experiment was carried out in triplicates and expressed in graph as mean ±SD of cell viability.
2.12.Analysis of activation of MAPK and AKT pathways following intracellular delivery of MAPK siRNA with selected Sr particles
Following 48 h treatment,the cells (MCF-7 and 4T1) treated with MAPK-siRNA in free and particle bound forms (SrSO,SrSOand SrF) were individually lysed with IP lysis buffer.The lysates were centrifuged at 13 000 RPM for 20 min at 4°C.5μl of the supernatant comprising soluble proteins was collected to estimate the total amount of proteins through bovine serum albumin (BSA) assay kit based on the manual.In the initial step,BSA protein was used to create the standard curve,which was to calculate the total protein concentration of cellular lysates based on their absorbance intensity.The remaining samples were aliquoted and stored in −80°C for subsequent SDS-PAGE and Western blotting.The cellular lysates containing 30μg of total protein were mixed with 10μl of 10X loading dye and subjected to SDS-PAGE using stain free mini protein SFX gels (15 wells) in 1X running buffer at 0.01 amp/gel.7μl precision plus protein standards-dual color was used as molecular weight marker to establish the molecular weight of the sampled protein.Transfer of protein samples from gel to the 0.2μm PVDF membranes attached to trans-blot turbo transfer pack through trans-blot turbo transfer system was performed for 7 min at 1.3 amp,followed by blocking in 5% skimmed milk in 1 × TBST for 1 h at room temperature.The transferred membrane was incubated with primary antibodies (pMAPK,TMAPK,pAKT,TAKT and GAPDH as loading reference) at 4°C overnight with gentle shaking,followed by washing in 1X TBST 5 times to remove unbound primary antibodies.Horseradish peroxide(HRP)-conjugated goat anti-rabbit secondary antibody Ig G(1:3000) was introduced into the membrane for 1 h with mild agitation,before washing for 5 times in 1 ×TBST to again eliminate the inbound antibodies.The membrane was exposed to the mixture of enhanced chemiluminescence(ECL) for 5 min before observation of bands through chemiluminescence signals using XRS Chemidoc system(Bio-Rad,USA).
3.Results and discussion
3.1.Fabrication and screening of inorganic Sr particles
3.1.1.Turbidity measurement and optical microscopic images of Sr salt particles
Direct mixing of SrClwith NaSO,NaSO,NaF,NaCOor NaHPOresulted in formation of particles,accompanied by nucleation and growth,as reflected by an increase in the turbidity measured at an wavelength of 320 nm (Fig.1 A).Optical microscopic images were also captured for the particles formed in the same way (Fig.1 B).SrSOdemonstrated greater number in particle formation with possibly large-sized particulates,followed by SrSOand Sr(PO) 2 .SrFand SrCOwere comparable with COAP in growth.Images seen under the microscope also revealed high number of SrSOand SrSOparticles,suggesting greater driving force for particle formation.Generation of inorganic strontium salt particles is highly dependent on pH,concentrations of reactants,and time and temperature of incubation.Particle formation accelerates as the concentration of reactants increases,acting as a driving force for the chemical reaction [36].
3.1.2.Effect of reactant concentration on Sr salt particle formation
The association between concentration of each reactants with turbidity (Fig.2 A) revealed that particle formulation with 2 mM of strontium salts exhibited slower particle growth and less generated aggregates compared to 5 and 10 mM,suggesting the growth is dependent on the reactant concentrations when time and temperature of incubation and pH are constant.As the cation-providing salt,SrClconcentration increases,(with constant amount of anion-providing salt),the rate of precipitation reaction rises.Particle synthesis activity was also accelerated with higher concentrations of anionproviding salts (fixed SrClconcentration (Fig.2 B).Nucleic acid binding efficiency of a carrier is highly associated with particle size and number,where excessive formation may lead to larger formed aggregates,preventing effective formation of gene loading-particle complexes,and subsequently hinders cellular uptake [37].There was relatively small difference between the turbidity readings for 2 and 5 mM cation-providing salts,which prompted the selection of 5μl of 1M SrCland 2μl of 1M anion-providing salts for subsequent experiments.Greater cationic domains from 5 mM Sr were needed for a stronger attraction with negatively-charged nucleic acid,resulting in superior binding activity [38].

Fig.1–Turbidity measurement and optical microscopic images of various Sr salt particles.(A) Absorbance intensity of Sr particles generated via precipitation reaction.Addition of 5μl of 1M SrCl2 into 5μl of 1M Na2SO4,Na2SO3,NaF,Na2CO3 or Na2 HPO4 to generate various salt crystals upon 30 min of incubation at 37°C.Serum-supplemented DMEM media was added to achieve 1 ml strontium particle suspension.Absorbance at 320 nm wavelength was measured for all fabricated particles using spectrophotometer with reference to CO3 AP.(B)Microscopic observation of strontium particles.Images were captured at 10 ×resolution.
3.1.3.Effect of incubation time,pH and temperature on Sr salt particle formation
Elevation in pH,temperature and incubation time generally shifts the reaction equilibrium towards the forward direction by enhancing ionization and diffusion of the reactants.Particle formation was greatly enhanced with longer incubation period (Fig.3 A).Turbidity of salt particles was seen at 0 min incubation,revealing that particles formed immediately upon mixture between two soluble salts.The growth continued at 60 min,with at least 0.3 difference of absorbance values from the initial time.pH of HEPES buffer(Fig.3 B) and incubation temperature (Fig.3 C) under fixed concentration of the reactants are the physical factors that results in increment in energy level and boosting the rate of particle reaction [39,40].Formation of Sr salt particles was even seen at 4°C,although the rate was relatively slow.Incubation at 60°C resulted in more particle aggregation associated with the boost in kinetic energy.Similarly,alkaline pH of HEPES was associated with greater rate of particle generation.Higher rate of particle formation often leads to larger particle sizes [41,42].Turbidity analysis revealed that SrSOand SrSOdemonstrated fastest particle growth,while SrFwas slowest,resembling COAP activity.
The association of greater particle growth with formation of larger particles was further determined via estimation of the average diameter for each Sr particles using ZetaSizer.The zeta potential revealed an inclination of more negative particles with higher concentration of anion-providing salts.
3.1.4.Analysis of size and zeta potential of generated Sr salt particles
Turbidity and particle size measurement both verified that precipitation reaction of Sr salts are driven by the concentration of soluble salts,reactant pH,the incubation temperature and time to generate a supersaturated solution,leading to particle nucleation into matured crystals of various sizes.SrSOand SrSOexhibited particles in higher number and larger sizes,ranging from 471 nm to 721 nm in diameter (Table 1 ).SrSOand SrSOformed particles were larger in comparison to COAP particles.SrFparticles were comparatively smaller with the average size of 105 nm (at 2 mM of NaF).Particles with size range of 20—200 nm have a greater tendency to accumulate and be retained within the tumor environment due to the existence of leaky vasculature and poor lymphatic systems,respectively,within the tumor microenvironment [40].Larger particles are susceptible to non-specific accumulation to other organs,while smaller ones with size less than 5 nm have low circulation half-life due to greater tendency for renal elimination [43].Higher circulation time associated with small particle sizes of Sr salts enhances particle buildup on the tumor site to promote claritin-associated cytoplasmic internalization [35].Particles with < 200 nm in diameter is therefore ideal for transporting genetic materials into the cells via receptormediated endocytosis [42].

Table 1– Particle size and zeta potential analysis of Sr particles.Fabricated Sr particles were analysed using Zetasizer,with reference to CO3 AP.
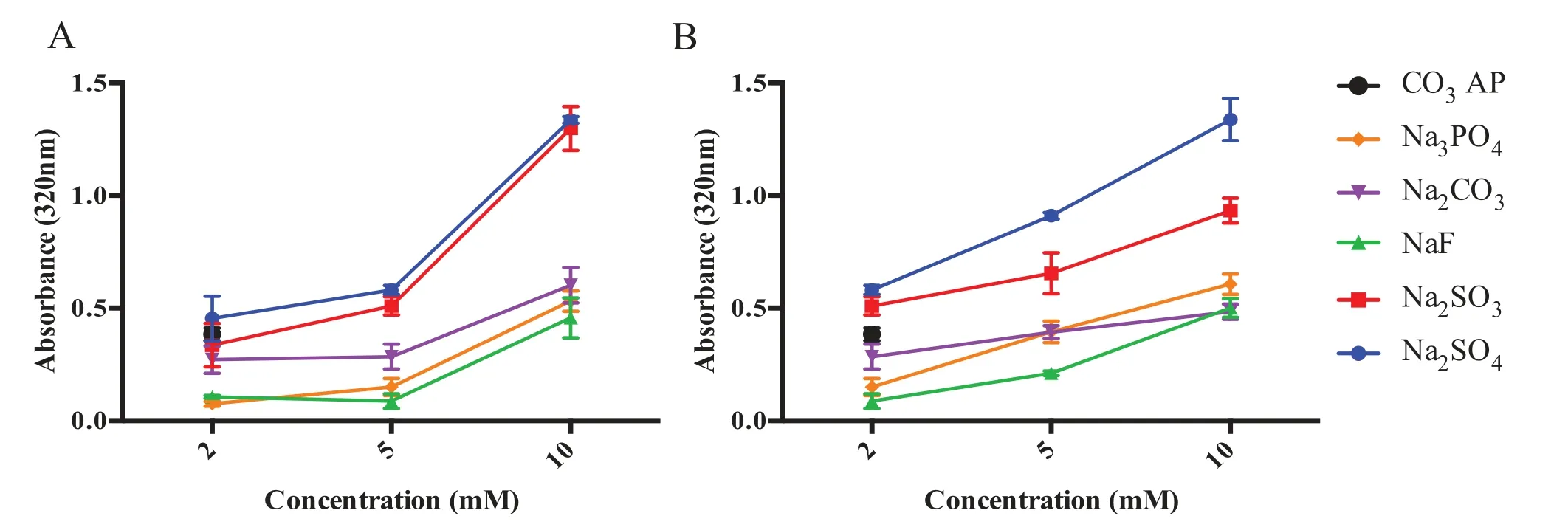
Fig.2–Effect of salt concentration on various Sr particle formation.(A) Influence of cation-providing salt concentration on particle formation.Various concentration of SrCl2 was introduced into 10μl HEPES-buffered media (pH 7.5),followed by 5μl of 1M Na2SO4,Na2SO3,NaF,Na2CO3 or Na2 HPO4,generating Sr salt particles upon 30 min of incubation at 37°C.(B) Influence of anion-providing salt concentration on particle formation.5μl of 1M SrCl2 was introduced into 10μl HEPES-buffered media (pH 7.5),followed by mixing of different concentration of Na2SO4,Na2SO3,NaF,Na2CO3 or Na2 HPO4 (2,5 and 10μl of 1M).Subsequently,FBS-containing DMEM media was added to achieve final volume of 1 ml Sr particle suspension.Absorbance at 320 nm was measured for all fabricated Sr particles using spectrophotometer with reference to CO3 AP.
Negative surface charge ranging from −6 to −11 mV was measured for all Sr salt particles.Zeta potential is an electric potential of particles,associated with particle stability and surface morphology [44].However,measured surface charges were not associated with intrinsic charge present on the particles,as the environment of the solution containing different ions would significantly influence the average zeta potential.Strontium ions in the particles contribute to the binding with negatively-charged nucleic acids and anionic cell surface [45].
3.1.5.Estimation of nucleic acid binding affinity to various Sr salt particles
Adsorption of DNA to Sr salt particles was assessed by linking PI,a fluorescence dye to the DNA through intercalation,and thereby forming pDNA-labeled Sr salt particles.Fluorescence imaging demonstrated fluorescent emission predominantly from the labeled SrF,SrSOand SrSOcomplexes.Indirect quantitative assay based on estimation of free (unbound)PI-labeled plasmid present in the supernatant (following centrifugation of various Sr salt particles) (Fig.4 B) indicates that SrFhas similar binding capacity with COAP,at approximately 90%,while SrSOand SrSOrevealed 80%binding towards the pGFP.siRNA binding affinity were investigated using fluorescence labeled AF488 siRNA.SrSOdemonstrated highest percentage of siRNA binding,with> 90% AF488 siRNA bound,followed by SrFand SrSO(Fig.4 C).The least binding interactions between pGFP or AF488 siRNA and Sr particles were seen for SrCOand Sr(PO) 2 complexes.COAP had high electrostatic affinity towards the nucleic acids,allowing efficient transportation of genesloaded COAP across the plasma membrane [42].Weak intensity seen with PI-tagged particles (Fig.4 A),especially COAP may be associated with existence of tiny particles emitting faint fluorescence signals from the relatively small number of pDNA bound to a particle.
3.1.6.Analysis of cellular uptake of pDNA and siRNA mediated by Sr salt particles
Cellular uptake of nucleic acid-loaded particles complexes was greatly affected by both nucleic acid binding capacity and the average particle sizes of Sr complexes [36].Low binding affinity of a carrier may affect the amount of gene transported into the cells via gene-carrier complexes.Internalization of smaller-sized particles by cells may increase up to 20-folds in comparison to larger ones [40].Fluorescence microscopic observation following 4 h incubation of MCF-7 cells with fluorescent pGFP and AF 488-labeled Sr salt particles revealed significant amount of genetic materials either associated with the extracellular signaling or internalization of the complexes via endocytosis,seen on both Fig.5 A and 5 B.Highly noticeable fluorescence signals were visualized for SrSO,on both genetic materials.Weaker signaling were seen with both SrCOand Sr(PO)particles,which may be reflected by their low affinity towards nucleic acids,as seen in the earlier Fig.4 .Fluorescence signaling were greater with AF 488 siRNA in comparison to pGFP,possibly associated with smaller size of siRNA that enables stronger adherence towards Sr particles,resulting more siRNA-bound Sr complexes to adhere strongly towards the cellular membrane to initiate cell-mediated endocytosis.Large ionic molecules cannot easily cross the phospholipid bilayers of the cell membrane,thus required endocytosis process for the complexes to be internalized.The surface charge of the particle complexes may influence the ionic interaction with the negatively-charged syndecans of the cellular surface.In the absence of cationic domains as seen in naked genes,internalization were ineffective as the result of repellent action of both negative domains [45].In summary,both gene binding and cellular uptake activity revealed greater activity with SrSO,SrSOand SrFNPs as compared to SrCOand Sr(PO) 2 particles,with less than 60% binding with latter NPs.Subsequent experiments were therefore focused on the activity of three said salts for greater possibility of effective gene transfection.
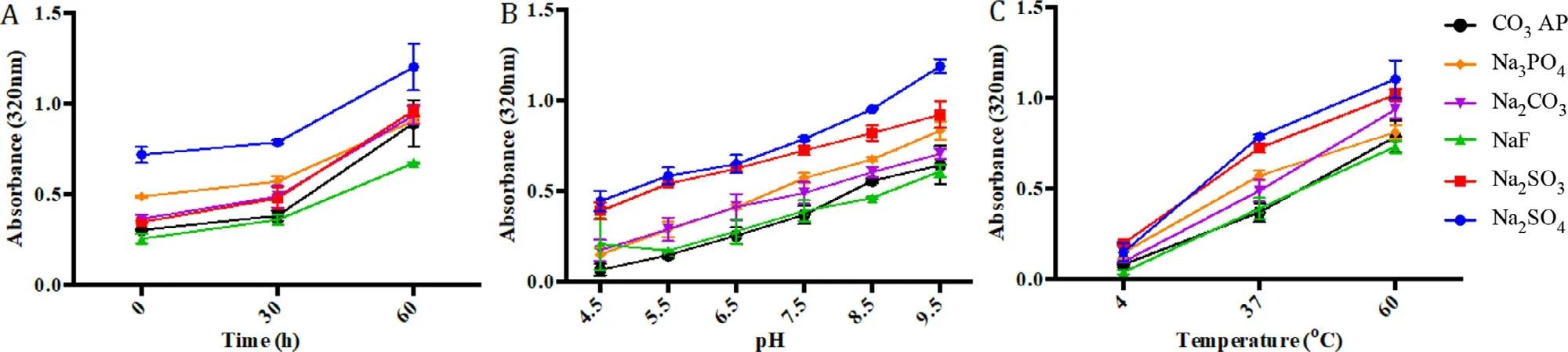
Fig.3–Effect of different variables on Sr particle formation.(A) Influence of incubation time on particle formulation.5μl of 1M SrCl2 was introduced into 10μl HEPES buffered media (pH 7.5),followed by missing of 2μl of 1M Na2SO4,Na2SO3,NaF,Na2CO3 or Na2 HPO4,generating various Sr particles upon incubation at 37°C at varying time points (0,30 and 60 min).(B)Influence of pH adjustment of Sr particle formulation.5μl of 1M SrCl2 was introduced into 10μl HEPES buffered media (pH ranging from 4.5 to 9.5),followed by mixing of 2μl of 1M Na2SO4,Na2SO3,NaF,Na2CO3 or Na2 HPO4,generating various Sr salt crystals upon 30 min incubation at 37°C.(C) Influence of incubation temperature on particle formulation.5μl of 1M SrCl2 was introduced into 10μl HEPES buffered media (pH 7.5),followed by mixing of 2μl of 1M Na2SO4,Na2SO3,NaF,Na2CO3 or Na2 HPO4,generating various Sr salt crystals at 37°C upon various incubation time.Subsequently,FBS containing DMEM media was added to achieve final volume of 1 ml particle suspension.Absorbance at 320 nm was measured for all fabricated Sr particles,with reference to CO3 AP.
3.2.Visualization of Sr particles size and morphology via FE-SEM
Microscopic visualization of Sr particles were obtained to understand the shape and morphology of the particles.Images of the selected Sr particles (SrSO,SrSOand SrF) via FE-SEM demonstrated that fabricated SrSONPs are relatively spherical while SrFNPs were seen as clusters of rods(Fig.6 ).The characteristics of the particle shape may influence the subsequent cellular adhesion and internalization,as greater activity was mostly noticed with spherical salt crystals,associated with greater surface areas for cell adhesion.Nanorod particles are known to possess greater cell surface adherence efficiency,[46]which might have helped attachment of gene-SrFcomplexes during the cellular uptake studies.Slight variation in the images seen in comparison to Zetasizer measurement (Table 1 ) was likely due to the tendency of particles to aggregate during drying step and imaging procedure that may disrupt the hydrodynamic diameter of SrNPs [41].Morphology of nanoparticles is in fact one of the major determining factors for efficient cellular uptake [47].
3.3.Determination of cytotoxic activity of Sr particles
One of the fundamental prerequisites for clinical applications of a nanocarriers is that it should be nontoxic to human cells.Studies on both MCF-7 and 4T1 cells at three different time points (24,48 and 72 h) were conducted,where Sr salt particles were found to cause transient increase in viability in both cells lines at 24 h,ranging from 110% to 140%,with viability remained high on Day 3,ranging from 90% to 95%,as shown in Fig.7 .Cellular proliferation following treatment was greater in MCF-7 as compared to 4T1 cells.Fiona et al.described the ability of SrClsalt to trigger the release of endothelial growth factor,causing activation of RhoA/Rac1 which is associated with osteoblastogenesis enhancement and adipogenesis limitation [42].Sr+ is known to promote proliferation and differentiation in cells that requires high concentration of calcium to facilitate their physiological processes such as growth and differentiation.Introduction of Sr enables the cells to compensate the lack of calcium to facilitate their survivability as observed in human bone marrow stromal and mesenchymal stem cells [48,49].Despite that,cellular growth was comparable with COAP particles upon 48 h of incubation,suggesting their reasonable safety level for subsequent cellular studies.
3.4.Dissolution activity of Sr particles in acidic environment

Fig.4–Binding affinity of genetic materials towards Sr particles.(A) Fluorescence microscopic observation for binding affinity of pDNA towards Sr particles.5μl of 1M SrCl2 was introduced along with PI-stained p53 (1:1 ratio) into 10μl HEPES buffer media,followed by mixing of 2μl of 1M Na2SO4,Na2 SO3,NaF,Na2CO3 or Na2 HPO4 and incubation for 30 min at 37°C.FBS-containing DMEM media was added to achieve 1 ml solution.Image was captured at 10 ×resolution under PI filter,with reference to CO3 AP.(B) Fluorescence analysis for binding affinity of pDNA towards the Sr particles formulated in presence of pGFP as described earlier.(C) Fluorescence analysis for binding affinity of AF488 siRNA towards Sr particles.Quantitative measurement of pDNA/siRNA bound particles was achieved with multi-label plate reader following centrifugation of fabricated particles at 15 000 RPM for 5 min and aspiration of 100μl resuspended solution into 96 well-plate,prior to fluorescence intensity measurement.
Investigation on pH-responsive characteristic of selected Sr salt NPs was performed through exposure of NPs in the acidic environment to mimic the late endosomal stage.The later endosome has pH of approximately 3.5—4.5,where COAP was able to disintegrate and to release pDNA via proton sponge effect [44].Dissolution of the particles within endosomal cavity should lead to accumulation of cations and anions,causing high osmoticity within the cavity,resulting in the swelling and rupturing the NPs.Dissolution of Sr salts upon exposure of acidic environment was conducted by adjusting the pH in the complexes (Fig.8 ) from the initial pH of 7.5 (which the particles were initially formed).Spectrophotometric reading revealed effective dissolution of SrSOand SrF,as absolute particles dissolution occurs at pH 4.5,seen by 0.0 in absorbance intensity on both salts.SrSOparticles demonstrated a decrease in absorbance value of 0.2 upon a drop in pH,which is relatively smaller than other Sr salts and COAP particles.These findings indicate that the salt particles were not able to be fully dissolved upon changing in pH,which might hinder the loaded genetic content to be completely released from the complexes.Response to changes in pH is therefore vital to prevent inefficient escape of pDNA and siRNA,partial or incomplete particle dissolution may impede effective transfection,resulting in lower transgene expression or inefficient silencing of an endogenous gene[44].It is expected that the salt particles would be able to maintain their structures especially in the blood which is slightly basic,with pH range of about 7.35—7.45.With the pH ranging from 7.0—7.4 in average physiological tissues,the salt particles should also not disintegrate prematurely to release their genetic load,thus promoting the gene activity(expression or silencing) particularly in the targeted cancer cells [50,51].
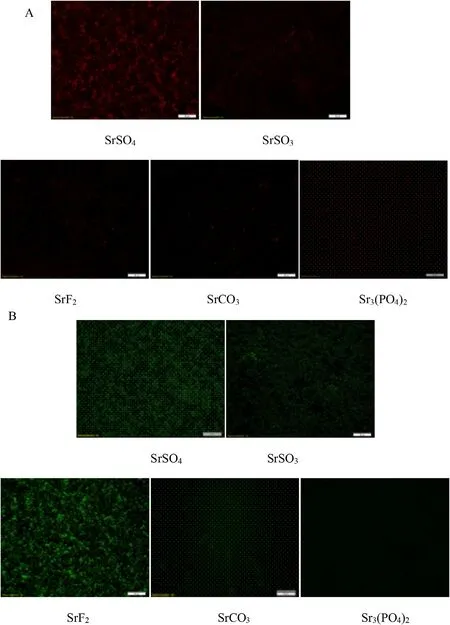
Fig.5–Fluorescence microscopic imaging of cellular uptake with Sr salt particles.Fluorescence activity emitted by MCF-7 uptake of (A) pGFP and (B) AF 488 siRNA-loaded Sr particle complexes were visualized.PI-labeled onto pGFP at the ratio of 1:1,prior to loading into various Sr particles.AF 488 siRNA loaded with different Sr particles were transfected onto the cells seeded at 50 000 in each well of a 24-well plate,followed with incubation for 4 h.Images were captured upon washing with EDTA in PBS at 10 ×resolution.
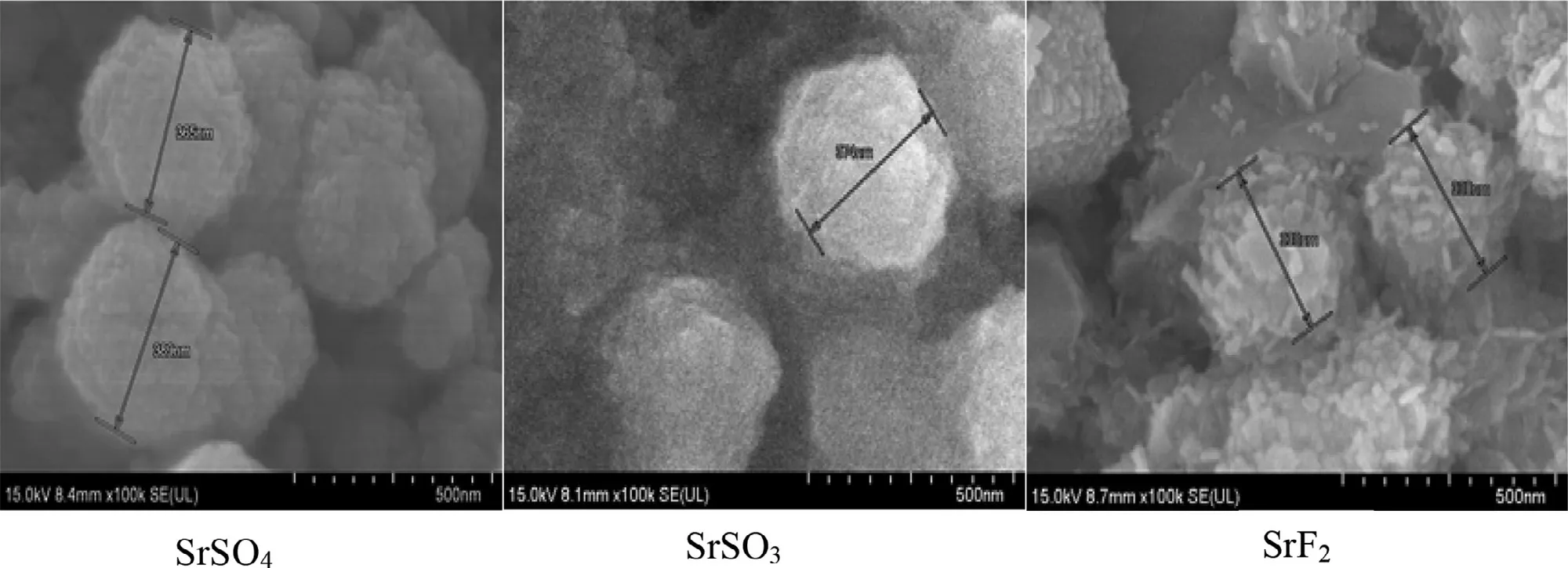
Fig.6–FE -SEM visualization of SrSO4,SrSO3 and SrF2 particles.Fabricated Sr particles were kept on ice prior to microscopic observation.1μl of resuspended solution was placed onto carbon tape-coated sample holder and dried at room temperature,followed by platinum sputtering of each salt samples for 60 s.Sputtered Sr samples were observed at 10–15 kV.

Fig.7–Cytotoxicity of selected Sr salts on mammary carcinoma cells,(A) MCF-7 and (B) 4T1 cells.50 000 of MCF-7 or 4T1 cells were seeded,treated with NPs and incubated for 24–72 h,with media substitution following first 4 h of incubation.50μl of MTT was incorporated into the treated cells after incubation,where the media containing MTT aspirated after 4 h incubation and finally addition of 300μl DMSO.Spectrophotometric reading of viable cells was observed at 595 nm wavelength with reference of 630 nm.Each selected inorganic Sr particles was individually compared with CO3 AP.
3.5.Transgene expression activity of Sr particles
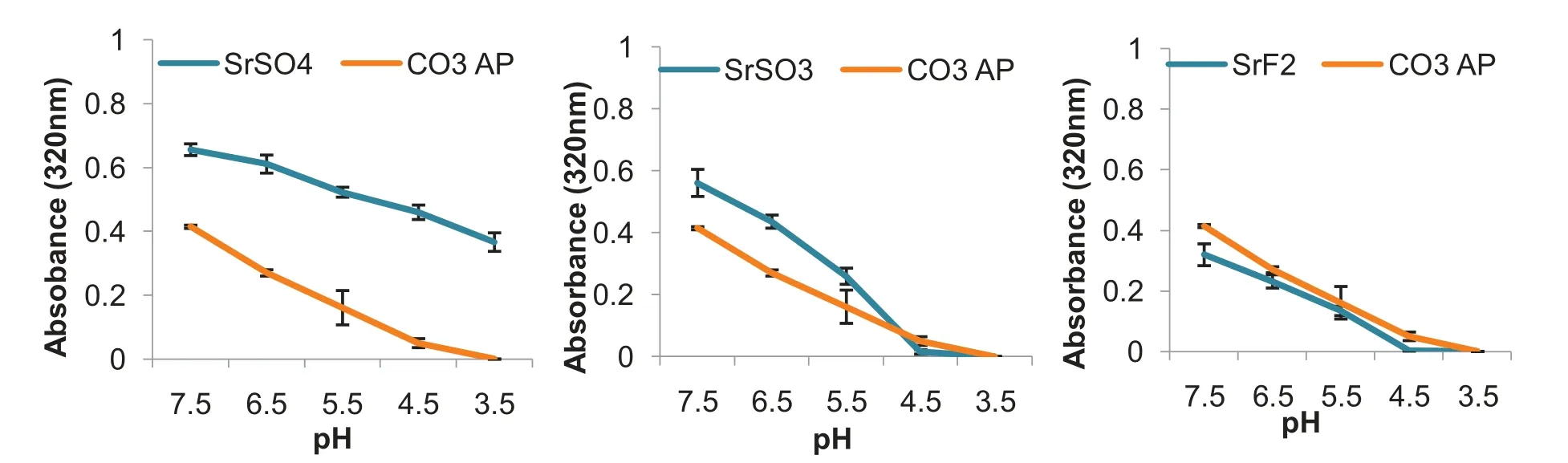
Fig.8–Dissolution of selected salt particles at acidic pH.HCl was incorporated into the prepared particle suspension following incubation at 37°C for 30 min to achieve pH of 6.5,5.5,4.4 and 3.5.Spectrophotometric reading of suspension was observed at 340 nm wavelength.
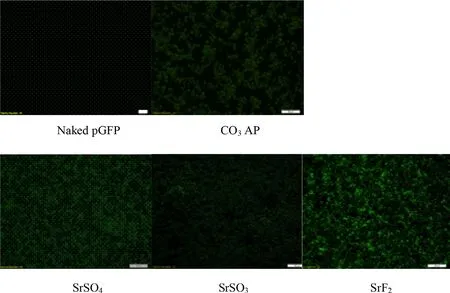
Fig.9–Fluorescence microscopic images of gene expression activity of pGFP-loaded Sr particles on MCF-7 cells.Each pGFP-loaded strontium complexes,SrSO4,SrSO3,and SrF2 in addition to naked pGFP and CO3 AP as control,was transfected into seeded MCF-7 cells,subsequently incubated for 4 h and washed with 10 mM EDTA in 1 ×PBS,incubation for additional 48 h,followed by observation under FITC-filtered fluorescence microscope.
High affinity for nucleic acids with subsequently high cellular uptake does not guarantee the endocytosed genetic materials transported via carrier will be adequately expressed or able to silence a target mRNA.Efficient transgene expression or knockdown of an endogenous gene requires a swift release of plasmid and siRNA from the endocytosed Sr salt complexes before being transported to the final destinations,nucleus and cytosol,respectively.Reporter gene expression is an indirect,but a powerful approach to assess the efficiency of a carrier in facilitating delivery and transcription of a desirable transgene in nucleus [52].The nucleic acid bound towards particles should be discharged before the content were subjected to the intercellular lysosomes for lysis or transported extracellularly via transcellular pump within the cellular membrane.In order to stimulate the fluorescence signals,pGFP has to pass through the nuclear membrane to initiate gene transcription,followed by translation of mRNA within the cytoplasmic proteins.Superior transfection activity of SrFwas demonstrated by highly-intensed fluorescenceexpressing cells,proposing pGFP release from the salt prior to the initiation of lysosomal and transcellular activity (Fig.9 ).Maximum emission of cellular luminescence activity was also identified with pGL3-loaded SrF,shown with 10 × greater RLU/mg protein in comparison to SrSOand COAP (Fig.10 ).Highly-intensified luminescence activity further revealed the effectiveness of SrFto allow effective DNA unloading and escape from the salt complexes,associated with pHdependent effective salt disintegration.SrSOand SrSOalso showed clear transgene expression activity although less superior as compared to SrF.Nonetheless,all three Sr salt particles studied exhibited better cellular activity than COAP.Naïv e pDNA deli v er y w as inefficient as seen in both fluorescence imaging and luminescence activity which may be associated with low internalization due to inability to adhere to cellular membrane and stimulate endocytosis process.
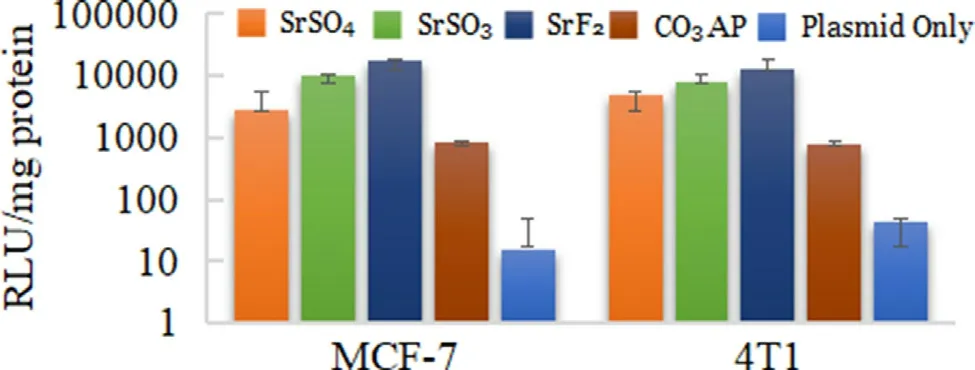
Fig.10–Luminescence intensity of pGL3-complexed Sr salt particles treated onto MCF-7 and 4T1 cells.Each type of pGL3-loaded selected salts,SrSO4 and SrSO3,was transferred into prepared wells containing seeded 50 000 MCF-7 or 4T1 cells and incubated for 48 h,with serum-supplemented media substitution following first 4 h of incubation and treatment with 5 mM EDTA in 1 ×PBS.Transfected cells were lysed after the removal of media,followed by lysate centrifugation at 15 000 RPM at 4°C for 10 min.100μl supernatant was aspirated to estimate the relative luminescence activity/mg of protein.
3.6.Cytotoxicity assessment following intracellular delivery of p53 and MAPK siRNA using selected Sr salt particles
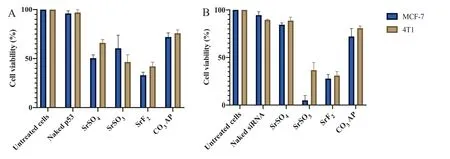
Fig.11–Cytotoxicity observation following introduction of genetic materials with SrSO4,SrSO3 and SrF2 particles.Cell viability of MCF-7 and 4T1 cells following treatment with p53-loaded Sr complexes (A).5 mM SrCl2 was mixed with 500μg of p53 prior to incorporation of 2 mM Na2SO4,Na2SO3 or NaF in 10μl of HEPES solution.Cell viability of MCF-7 and 4T1 cells after treatment with MAPK siRNA-loaded barium salt particle complexes (B).5 mM SrCl2 was mixed with 10 nM MAPK siRNAs prior to incorporation of 2 mM Na2SO4,Na2SO3 or NaF in 10μl of HEPES solution.Each set of experiments included untreated cells,CO3 AP and unloaded salt particles as controls.Nucleic acid-loaded complexes were transferred into each well of a 24-well plate containing cells seeded at 50 000 cells/well on the day prior.The cells were incubated for 48 h after replacement of particle-containing media with serum-supplemented media following first 4 h of incubation in presence of the various particles and subsequent treatment with 5 mM EDTA in PBS.Finally,50μl of MTT was incorporated into the treated cells,and the media containing MTT was aspirated after 4 h incubation.300μl DMSO was added to dissolve the formazan crystals.Spectrophotometric reading of viable cells was taken at 595 nm wavelength with reference to 630 nm.
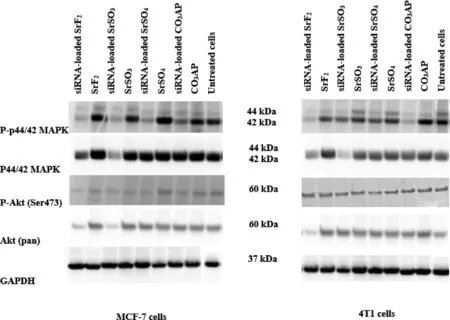
Fig.12–Protein expressions following treatment of MCF-7 and 4T1 cells with MAPK siRNA-loaded SrSO4,SrSO3 and SrF2 particles.Proteins obtained from lysates of treated cells were run on SDS-PAGE and transferred to PVDF membrane,followed by incubation with primary antibodies raised in rabbit against phosphor-p44/42 MAPK,p44,42 MAPK,phospho-Akt (Ser473)and Akt (pan).HRP-conjugated goat anti-rabbit secondary antibody was used to detect the chemiluminescence signals.Predicted bands for pMAPK,TMAPK,pAkt and TAkt are at 44,42 and 60 kDa,respectively.GAPDH was used as loading marker with bands achieved at 37 kDa.
Determination of the efficacy in transgene expression level and desirable particle sizes enables us to select three strontium salt particles,SrSO,SrSOand SrFfor further investigation on inducing cytotoxicity by facilitating intracellular delivery of p53 plasmid and MAPK siRNA.p53 is a prominent tumor suppressor with potential ability to arrest and kill carcinoma cells,whereas MAPK is a key enzyme of MAPK/ERK pathway responsible for ensuring proliferation of the cancer cells and therefore,silencing of MAPK expression with a specific siRNA could significantly inhibit the uncontrolled cell division [51].Target gene,p53 is a central regulator of cellular growth,DNA repair,and apoptosis,often down-regulated in cancer patient due to gene missing or malfunction.The introduction of wild-type p53 is hence necessary to normalize the regulatory mechanism of ‘guardian of genome’ gene [52,53].Both intracellular delivery of loaded p53 into SrFand SrSOparticles caused notable cytotoxicity in both MCF-7 and 4T1 cells (Fig.11 A).SrF-mediated delivery of the gene resulted in remarkably high toxicity in both cell lines,indicating efficient transgene expression following cytoplasmic release of the plasmid from the particles as well as from the endosomes,and thereby suggesting SrFparticles would serve as a powerful tool for effectively delivery of gene-based therapeutics.Activity of SrSOparticles was superior to SrFin promoting cell deaths both in MCF-7 and 4T1 cells through intracellular delivery of MAPK siRNA (Fig.11 B).Sr salt particles like other successful delivery vectors,e.g.,COAP are able to bind to the anionic cell membrane via ionic interactions through their cationic salt components (Sr),resulting in endocytosis and subsequent degradation of salt particles at low endosomal acidic pH at the late endosomic phase.DNA or siRNA can therefore be released and subjected to endosomal escape according to‘proton-sponge’ hypothesis [54,55].
3.7.Analysis of activation of MAPK and AKT pathways following intracellular delivery of MAPK siRNA with selected Sr particles
MAPK and AKT pathways are known to play key roles in cancer cell proliferation and survival,respectively,and both pathways can cross-talk to each other [56,57].Therefore,investigation on the activation levels of the two pathways following intracellular delivery of MAPK siRNA with the help of SrSO,SrSOand SrFparticles were performed.As shown in Fig.12,MAPK siRNA-loaded SrFand SrSOparticles enabled greater knockdown activity of MAPK and AKT,seen by the diminished band intensity,leading to inhibition of their phosphorylation to a distinctive extent in both cell lines,similarly to the silencing outcome seen with MAPK-loaded COAP particles.The capacity of siRNAmediated knockdown of Sr particles might be associated with their small diameter and high affinity towards nucleic acids,leading to prompt cellular uptake and subsequent acidic pH-dependent dissolution of the internalized particles in order to release the siRNA in cytosol [58].
4.Conclusion
SrFand SrSOnanoparticles have been proven to be the most potential nanocarriers for genetic material delivery into mammary carcinoma cells,particularly owing to their nanometer sizes,strong binding affinity for nucleic acids,efficient cellular entry and fast dissolution rate to allow swift escape of genetic materials from the particles upon exposure to acidic environment.Owing to their ability to react efficiently to pH changes,both Sr salts are able to maintain their structure at different environments in the tissues and blood circulation,ranging from pH 7 to 7.5.Both silencing of the target mRNA with a specific siRNA or translocating pDNA into the nuclear membrane for subsequent transcription and translation were therefore greatly enhanced.A pre-clinical study in an animal model of breast cancer will further elaborate the potential of Sr salt particles,especially SrFand SrSOfor intravenous delivery of nucleic acid-based therapeutics.
Conflicts of interest
The authors declare that there is no conflicts of interest.The authors alone are responsible for the content and writing of this article.
Acknowledgments
The authors acknowledge the financial support received from Ministry of Education Malaysia,for their support and encouragement in carrying out his college work.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi: 10.1016/j.ajps.2020.11.002 .
 Asian Journal of Pharmacentical Sciences2021年2期
Asian Journal of Pharmacentical Sciences2021年2期
- Asian Journal of Pharmacentical Sciences的其它文章
- Role of nanoparticle-mediated immunogenic cell death in cancer immunotherapy
- Control strategy and methods for continuous direct compression processes
- Heterobifunctional PEG-grafted black phosphorus quantum dots: “Three-in-One”nano-platforms for mitochondria-targeted photothermal cancer therapy
- Intra-Articular injection of acid-sensitive stearoxyl-ketal-dexamethasone microcrystals for long-acting arthritis therapy
- A computer-aided chem-photodynamic drugs self-delivery system for synergistically enhanced cancer therapy
- Gas-blasting nanocapsules to accelerate carboplatin lysosome release and nucleus delivery for prostate cancer treatment