
Genetic characteristics and molecular diagnostics of bone tumors
2021-07-20 01:10:48DavidSusterSaulSuster
David Suster ,Saul Suster
1Department of Pathology,Rutgers University,New Jersey Medical School,Newark,NJ 07103,USA.
2Department of Pathology,Medical College of Wisconsin,Milwaukee,WI 53226,USA.
Abstract The diagnosis of primary tumors of bone relies heavily on clinicopathological and radiological correlation and is often best performed in a multidisciplinary setting.Bone tumors comprise a heterogenous category of human lesions ranging from benign to malignant neoplasms.These tumors affect a wide age range and can become problematic for diagnosis when less common entities are encountered.Traditionally the pathological diagnosis of many bone tumors has been based primarily on the evaluation of hematoxylin and eosin-stained glass slides,sometimes combined with ancillary diagnostic techniques such as immunohistochemistry,conventional cytogenetics,fluorescence in situ hybridization,and polymerase chain reaction-based assays.More recently,the advent of massively parallel sequencing-based techniques has opened new avenues for diagnostic testing in bone tumors;however,these new testing modalities are sensitive to traditional decalcification procedures that are commonly used in the routine processing of bony specimens.Herein we provide a focused review concentrating on the molecular genetic features of bone tumors with specific,recurrent genetic alterations that make them appealing targets for directed ancillary testing by conventional or molecular techniques.In addition,specimen handling with regards to decalcification procedures are discussed and the different types of testing modalities available are reviewed.
Keywords: Molecular pathology,next generation sequencing,bone tumors,sarcoma,decalcification
INTRODUCTION
Primary tumors of bone comprise a heterogeneous group of benign and malignant tumors that affect patients of a wide age range.While benign bone tumors are relatively common,malignant bone tumors are exceedingly rare,accounting for less than 1%-2% of all neoplastic disease[1-3].While some of the more common bone lesions,such as osteochondroma,pose little problem for diagnosis,the less commonly occurring benign and malignant lesions can often lead to diagnostic difficulties.The histopathologic diagnosis of bone tumors is fraught with difficulty due to the histomorphologic overlap between many different types of tumors,including overlap between reactive,benign,and malignant lesions[4-5].In addition,the diagnosis of bone lesions is heavily reliant on correlation with imaging characteristics to be able to localize lesions to specific locations within different bones and to sometimes assist in staging[6-8].Thus,the diagnosis and treatment of patients with primary bone tumors is best accomplished with a multidisciplinary approach involving the coordination of surgeons,radiologists,pathologists,and oncologists to produce optimal patient care[9-10].
Given the inherent diagnostic difficulties associated with primary bone tumors,ancillary testing modalities that may assist in diagnosis are beneficial to patient care.Precision medicine for bone cancer has lagged behind soft tissue,epithelial,and hematologic neoplasms in part due to the special processing procedures required for bony tissues[11].Traditionally,diagnosis has relied on histopathological assessment of tumor tissue combined with clinical and radiological correlation.This has been supplemented with cytogenetic analysis including karyotype analysis and fluorescencein situhybridization (FISH)[12-16].Immunohistochemical analysis has played little role in the diagnosis of bone tumors,although recent molecular advances have provided pathologists with specific targets amenable to antibody interrogation in some tumors such as giant cell tumor of bone and chondroblastoma[17].Polymerase chain reaction (PCR)[18-20]and,more recently,massively parallel next generation sequencing (NGS) based assays have provided additional tools for assessing molecular alterations in bone tumors[21-23].However,it is worth noting that many of these ancillary testing modalities are exquisitely sensitive to conventional processing techniques,particularly decalcification,and care must be taken when processing bony lesions with consideration for downstream testing that may take place after the histopathological examination.This review aims to discuss the molecular genetic landscape of many of the primary bone tumors,specimen handling,and the ancillary testing available for diagnosis.
SPECIMEN HANDLING
Processing and handling are critical components for the proper diagnosis of bony surgical pathology specimens.When sufficient material is available,receiving fresh tissue is preferred as it allows for a portion of the tissue to be preserved (often fresh in saline or snap frozen) for subsequent cytogenetic,molecular,and microbiological studies[24].Priority however should be given to tissue that will be used for morphological analysis,as that remains the cornerstone of histopathologic diagnosis of bone lesions.Routine fixation using 10% formalin and paraffin embedding is acceptable for this purpose and generally does not interfere significantly with testing of antigen expression or genomic material[25,26].It is also worth mentioning that numerous other pre-analytical variables can have large effects on nucleic acid retrieval including specimen collection techniques,storage practices,temperature,duration of processing,and dehydration protocols amongst others.However,an in-depth discussion of pre-analytical variables is beyond the scope of this review which will focus on decalcification as it pertains specifically to bone specimens.Bone specimens often require additional preparation in the form of decalcification to allow for proper processing of the bone sections and creation of glass slides that can then be reviewed microscopically; however,decalcification is known to have detrimental effects on subsequent molecular testing[27,28].Decalcification is most often accomplished using strong (e.g.,hydrochloric acid or nitric acid) or weak (e.g.,acetic acid or formic acid) inorganic acidic solutions to aid in the demineralization ofbony specimens [Table 1].Although generally adequate for preserving histomorphology,decalcification with acidic solutions may lead to the degradation of genomic material and cause interference when molecular testing is performed[29,30].Acid decalcification with formic acid (a weak acid) has been reported to preserve genomic material for sequencing analysis although it can cause severe damage to DNA and RNA when longer decalcification times are used[31].Picric acid may also not be optimal for nucleic acid retrieval[32].Alternative methods or modifications to standard decalcification that exist include the use of ethylenediaminetetraacetic acid (EDTA),microwave,ultrasonography,and other types of acid.Acids or EDTA may be used in bone decalcification and combined with microwave or ultrasonography to reduce the time needed to decalcify,and EDTA decalcification alone appears to provide some measure of genomic material preservation compared to stronger acid solutions[33-37].Decalcification times vary for both acidic solutions and EDTA and can be modified with additional factors such as temperature or mechanical agitation[38].When handling bone specimens it is important to be familiar with the tissue processing procedures employed by the laboratory so that consideration can be given to the types of subsequent testing performed.

Table 1.Common methods of bone decalcification
DIAGNOSTIC TESTING MODALTIES
Karyotype,FISH,and chromosomal microarrays (CMA) such as array comparative genomic hybridization (aCGH)/single nucleotide polymorphism (SNP) arrays have all traditionally constituted the backbone of molecular diagnostics in bone and soft tissue tumors.In more recent decades,PCR based assays,firstgeneration sequencing technologies and the development of massively parallel targeted sequencing have provided new options for molecular testing of bone tumors[22,39-41].In general,there are certain advantages and disadvantages to each type of testing modality with some special considerations with regards to tissue processing.Karyotype and CMA,while less commonly used these days for bone tumors,require fresh tissue or perform better with fresh tissue and may still provide some valuable information when used.FISH,PCR,and NGS can be performed on formalin fixed paraffin embedded tissue; however,they suffer from degradation of genomic material during decalcification procedures particularly when strong acids are used.
The type of testing offered by different laboratories also varies widely making it important for pathologists and clinicians to understand the capabilities of each different type of assay and the information being returned.For example,FISH may be used to identify specific translocations or amplification of certain genes in a case where a particular diagnosis is suspected and can be done at a relatively low price with a rapid turnaround time; however,it is less sensitive than PCR or NGS and may lead to false negative results,particularly in bone tumors where neoplastic cells are present in the background of numerous benign cells[42].PCR and NGS based sequencing assays may be better diagnostic options for tumors where there is a broad differential such as a primary malignant small round blue cell tumor of bone,however these assays are exquisitely sensitive to decalcification and require proper triaging of pre-analytical variables such as tissue fixation and decalcification.In addition,NGS is expensive compared to older techniques and may not be available outside of larger academic institutions or referral centers.Table 2 provides a summary of some of the more common testing modalities that are available for diagnostic testing of bone tumors.
GENERAL MOLECULAR GENETIC LANDSCAPE OF BONE NEOPLASIA
Genetically bone tumors may be characterized in a similar fashion to soft tissue neoplasms,divided into lesions with simple genetics and lesions with complex genetics.These categories do not necessarily line up with the histological grade or clinical behavior of individual tumors; tumors with complex genetics are almost uniformly aggressive,although some tumors with simple genetics also behave in an aggressive fashion,and vice versa.Tumors that fall into the simple genetics category tend to have simple karyotypes and can generally be characterized by specific,recurrent molecular alterations that make them attractive targets for cytogenetic or molecular analysis.These alterations most commonly take the form of point mutations or chromosomal level abnormalities such as amplifications or translocations with various downstream effects such as the creation of oncogenic fusion proteins.Tumors from the complex category tend to display either multistep progression in their molecular profiles (such as high-grade chondrosarcoma and dedifferentiated chondrosarcoma) or are characterized by complex karyotypes with multiple molecular abnormalities present at the time of diagnosis (high-grade osteosarcoma and undifferentiated pleomorphic sarcoma).While the value of ancillary diagnostics is dependent on the aforementioned tissue processing protocols,when appropriate material is preserved,they can serve as a valuable tool in the diagnosis of difficult lesions that cannot be diagnosed on clinical,radiological,and histopathologic grounds alone.
Primary bone tumors represent a heterogenous group of tumors with a wide spectrum of differentiation.They include a variety of lesions from different categories including fibrogenic,osteogenic,chondrogenic,undifferentiated,and giant cell rich tumors.In addition,bone can be the site of various other tumors types including notochordal,vascular,myogenic and lipogenic tumors[3].A summary of the molecular alterations found in primary bone tumors is provided in Tables 3 and 4[43-121].While advances in molecular genetics have expanded our understanding of the molecular alterations present in many different types of tumors,the routine diagnosis of many primary bone tumors remains a clinicopathologic diagnosis rather than hinging on the identification of specific molecular alterations.As some of these lesions sometimes pose difficulty for diagnosis due to morphologic overlap with other lesions,they may benefit from ancillary molecular testing to identify specific alterations.In addition,a large variety of tumors that primarily occur within the soft tissues may rarely occur as primary osseous lesions.These tumors are mostly the subject of case reports or small series; however,they may cause problems for diagnosis when encountered as primaries outside of their usual site and many of them harbor specific genetic alterations that are helpful for diagnosis when encountered as a bone primary (see “Soft Tissue Tumors that Rarely Present as Primary Bone Tumors” section).Tables 3 and 4 provide a brief overview of selected lesions that exemplify the different categories of bone tumor genetics or in which molecular testing may be helpful.

Table 2.Cytogenetic and molecular diagnostic tests available for bone tumors
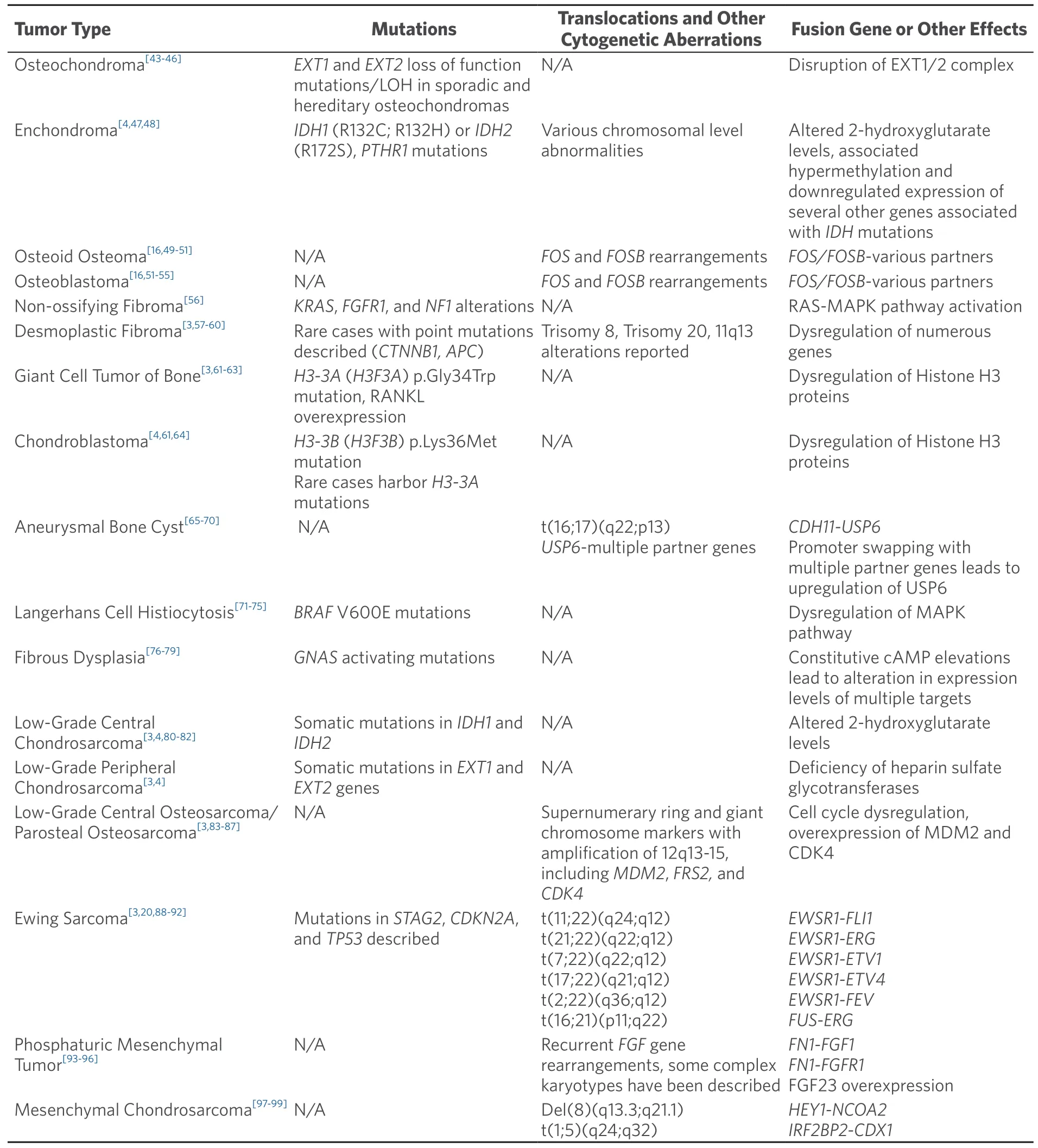
Table 3.Molecular genetic alterations in primary bone tumors (simple genetics)
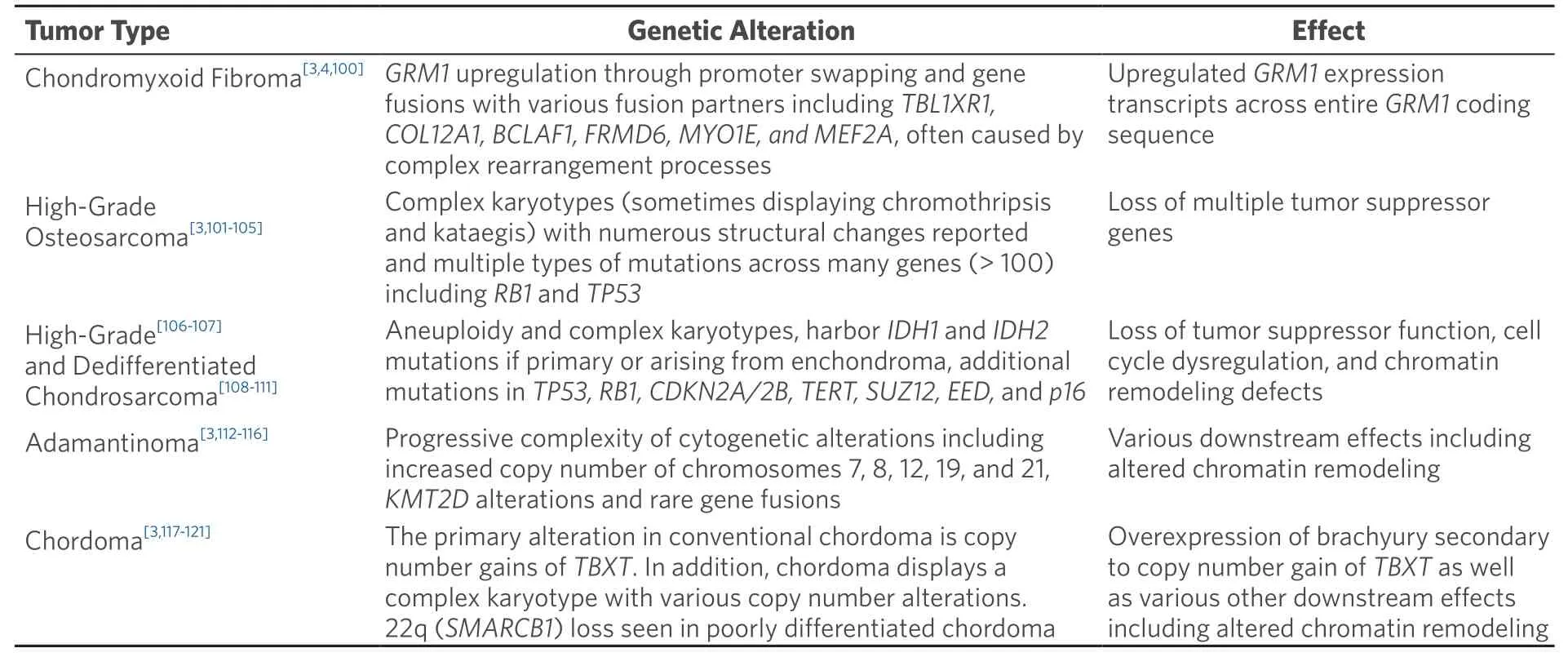
Table 4.Molecular genetic alterations in primary bone tumors (complex genetics)
MOLECULAR GENETICS OF SELECTED BONE TUMORS
Giant cell tumor of bone
Giant cell tumor of bone (GCT) is a benign primary bone tumor characterized by a mononuclear cell population of neoplastic cells with an admixed population of multinucleated osteoclast-type giant cells.These lesions fall into the simple genetics category of bone tumors.GCT represents approximately 5% of all primary bone tumors and may behave in a locally aggressive fashion[3].Recent molecular advanceshave identified that nearly all of these tumors are characterized by somatic point mutations in theH3-3A(H3F3A)gene leading to a specific amino acid substitution p.Gly34Trp that is detectable with immunohistochemistry (G34W IHC).This mutation leads to epigenetic modification and abnormal function of histone protein H3[61].In addition,studies have shown that other alterations including 20q11.1 amplification,IDHmutations (although this is disputed),and RANKL overexpression are present in GCT[62-64].While the diagnosis of GCT is generally straight forward some cases may display morphologic overlap with other giant cell rich lesions including chondroblastoma.For this reason,immunohistochemistry ormolecular testing for theH3-3Amutation may be useful in clinching the diagnosis.Achieving a correct diagnosis for these lesions is particularly useful as the tumors are amenable to targeted therapy with denosumab[64].
Chondroblastoma
Chondroblastoma is a rare benign primary bone tumor of young adulthood that displays chondrogenic differentiation and is part of the simple genetics category of lesions.It is characterized by a monotonous population of what appear to be primitive chondroblastic cells [Figure 1][4].These tumors are driven by mutations primarily in theH3-3B(H3F3B) gene,with an extremely small percentage harboring mutations inH3-3A[4,61].The mutations result in an amino acid change; p.Lys36Met that is detectable by immunohistochemistry (K36M IHC) [Figure 2][3,4,64].As in GCT,this mutation interferes with histone protein H3[61].Some cytogenetic abnormalities have been described including some rearrangements,although they do not appear to be recurrent[3].Chondroblastoma has also been shown to harbor RANKL overexpression similar to GCT and in rare cases has shown response to denosumab therapy in refractory tumors[64].Chondroblastoma may in some cases display morphologic overlap with both giant cell rich lesions as well as chondrogenic lesions.In some cases,prominent secondary aneurysmal bone cyst-like changes may be present[3],making the identification of the K36M mutation particularly useful.
Osteoblastoma and osteoid osteoma
Osteoblastoma and osteoid osteoma are two related lesions that are both osteoid producing primary bone tumors composed of immature woven bone spicules,prominent stromal vessels,giant cells,and prominent osteoblasts [Figure 3][3].Until recently molecular studies consisted of only a few cytogenetic studies that did not identify any recurrent alterations.More recently recurrent rearrangements ofFOSandFOSBhave been identified that in many cases can be identified with immunohistochemistry for FOS [Figure 4][16,50,51].FOSis a tightly regulated transcription factor that has been known to be involved in the pathogenesis of bone tumors;FOSandFOSBrearrangements involve multiple partners and create a mutant fusion transcript lacking the normal regulatory elements[16].A small subgroup of non-FOS-rearranged osteoblastomas have also been identified to be characterized by loss ofNF2[54].Osteoid osteoma in general i30s readily diagnosed on clinicopathologic grounds; however,osteoblastoma can present a problem for diagnosis as it may show overlap with some locally aggressive or outright malignant lesions such as osteosarcoma.While many osteoblastomas will showFOSimmunoreactivity,up to 14% of osteosarcoma samples also showed immunoreactivity in one study[51].This potential for non-specificity for osteoblastoma immunohistochemistry means that FISH or sequencing based assays are preferable as an ancillary molecular tool if fresh tissue is available; however,a positiveFOSimmunohistochemistry may help support a suspected diagnosis of osteoblastoma.

Figure 1.Chondroblastoma:high power magnification of chondroblastoma shows a monotonous appearing cell population with welldefined cell borders and ample eosinophilic cytoplasm.Some admixed multinucleated giant cells are present (Hematoxylin and Eosin,200x magnification)

Figure 2.K36M immunohistochemistry:immunohistochemistry for the K36M antibody shows a nuclear staining pattern within the neoplastic cells of chondroblastoma consistent with H3F3B mutation (K36M immunohistochemistry,20x magnification)

Figure 3.Osteoblastoma:low power magnification shows a highly vascular neoplasm with small spicules of immature woven production rimmed by osteoblastic cells (Hematoxylin and Eosin,20x magnification)
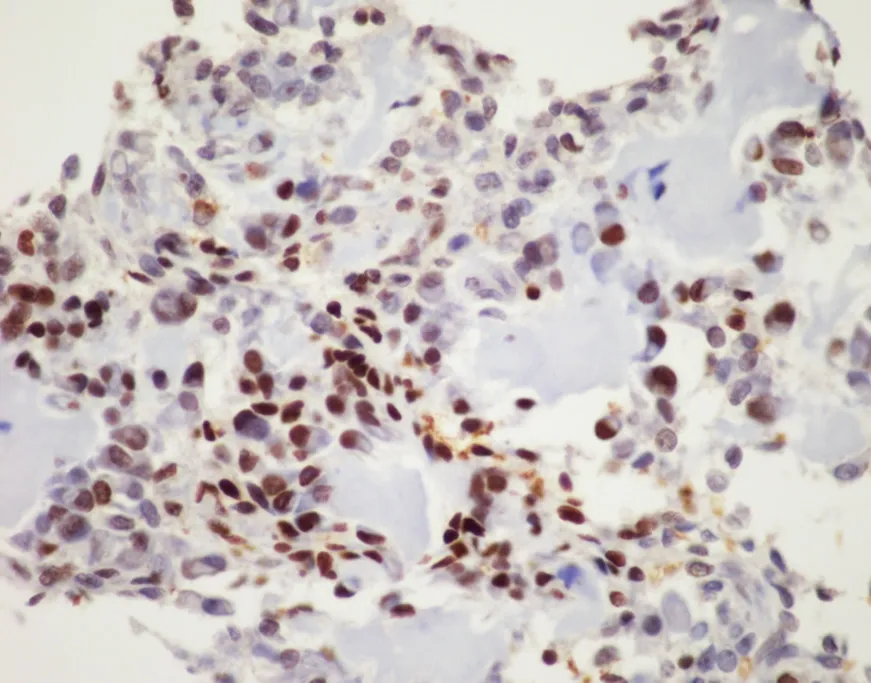
Figure 4.FOS immunohistochemistry:immunohistochemistry for FOS shows nuclear positivity for the majority of the neoplastic tumor cells in osteoblastoma,this serves as a surrogate for the presence of FOS rearrangements,although it is not entirely specific (FOS immunohistochemistry,20x magnification)
Aneurysmal bone cyst
Aneurysmal bone cyst (ABC) is a benign,yet destructive,primary bone lesion composed of multiple cystic blood-filled spaces that has a characteristic radiological impression with fluid-fluid levels[3].Cytogenetic characterization of these lesions has shown recurrent aberrations of chromosome 17p[65].These aberrations are now known to be recurrent rearrangements involving theUSP6gene[66].ACDH11-USP6rearrangement was the first described,although numerous different translocation partners have now been identified with a common mechanism involving promoter swapping with upregulation ofUSP6[66-70].The upregulation of USP6 leads to increased production of matrix metalloproteinases that lead to osteolysis,inflammation,and vascularization[66].USP6rearrangements are most easily identified using FISH break apart probes or next generation sequencing based fusion panels and are useful in the diagnosis of these lesions as there can be morphologic overlap between ABC and various benign and malignant lesions that can show secondary ABC-like areas.
Chondrosarcoma
Chondrosarcoma is defined as a malignant cartilage producing tumor and is comprised of a large family of tumors that includes multiple types such as conventional chondrosarcoma,periosteal chondrosarcoma,mesenchymal chondrosarcoma,and clear cell chondrosarcoma[3].The most commonly encountered of these entities is conventional chondrosarcoma which can be further classified into peripheral (located in the appendicular skeleton),central (located within the axial skeleton),primary (arising in the absence of a precursor lesion),or secondary (arising from a pre-existing bone tumor,usually enchondroma or osteochondroma)[3,4].Chondrosarcoma is graded based primarily on the degree of cytologic atypia and some other features including overall cellularity,cartilage matrix degeneration and mitotic activity.Lowgrade lesions occurring in the appendicular skeleton are termed atypical cartilaginous tumors.Genetically,primary peripheral and central chondrosarcomas are characterized by somatic point mutations in the isocitrate dehydrogenase genes,IDH1(R132C/H/G) andIDH2(R172S) leading to altered hydroxyglutarate levels[3,4].Secondary chondrosarcomas arising from enchondromas,a finding that often occurs as part of the genetic syndromes: Ollier disease and Maffuci syndrome,are also characterized byIDHmutations.Secondary peripheral chondrosarcomas arising in association with osteochondromas are characterized by mutations inEXT1andEXT2,the same genes mutated in patients with multiple hereditary exostosis[4,80-82].High-grade chondrosarcoma shows progressive molecular alterations including aneuploidy and complex karyotypes,and mutations inRB1,TP53,andCOL2A1[3].Finally,periosteal chondrosarcoma has been reported to harborIDHgene mutations while mesenchymal chondrosarcoma shows a characteristicHEY1-NCOA2rearrangement[3,98].Though some genetic abnormalities have been reported in clear cell chondrosarcoma its genetic profile has not been completely elucidated[3].Despite the presence of various molecular alterations in the different subtypes of chondrosarcoma it is worth noting that the diagnosis of conventional chondrosarcoma in general does not require ancillary molecular genetic testing and is instead established based on clinical,radiological,and histological criteria in routine clinical practice.However,associated molecular alterations may be useful in some situations,particularly in secondary lesions that may be related to germline conditions,when dealing with mesenchymal chondrosarcoma presenting as predominantly poorly differentiated small round blue cells,and in some cases separating conventional chondrosarcoma from other cartilaginous lesions when limited tissue is available for examination.
Osteosarcoma
Osteosarcoma is a malignant bone forming tumor that is the prototypical example of a primary bone sarcoma with complex genetics,but also includes some subtypes that are characterized by well-defined recurrent genetic alterations.These tumors are categorized by neoplastic osteoid production and a population of atypical osteocytes that can display a wide spectrum of cytological appearances from low-grade,bland appearing spindle cell lesions to high grade lesions with bizarre,atypical forms[3].The molecular genetics of high-grade osteosarcoma are complex; cytogenetic and large-scale sequencing studies have identified a number of somatic mutations as well as numerous,generally non-recurrent,copy number alterations at the chromosomal level[102-104].Somatic mutations in tumor suppressor genes and proto-oncogenes includeTP53,RB1,BRCA2,BAP1,RET,CDKN2APTEN,WRN,ATRX,and many others.Many of these genes such asTP53,RB1,andWRNare associated with hereditary cancer syndromes including Li-Fraumeni,hereditary retinoblastoma,and Werner syndrome,that predispose patients to an increased risk of osteosarcoma[102,103].Reported mutations are numerous and number in the hundreds[102].High-grade osteosarcoma is also characterized cytogenetically by chromothripsis (Greek origin,“thripsis” meaning shattering) and kataegis (Greek origin,“Kataegis” meaning thunderstorm) in which catastrophic chromosomal breakage occurs sometimes in combination with regional hypermutation that occurs through complex mechanisms [Figures 5 and 6][102,104].

Figure 5.Graphical schematic depicting chromothripsis:chromosomal shattering occurs with subsequent failure of genomic repair mechanisms resulting in inappropriate recombination of chromosomal fragments.During this process fragments of the chromosome may be lost or remain and reassembled erroneously
Low-grade variants of osteosarcoma include parosteal osteosarcoma and low-grade central osteosarcoma[3].The low-grade variants of osteosarcoma are characterized by a bland appearing spindle cell population of cells rather than the overtly malignant cells present in high-grade osteosarcoma [Figure 7].Both of these subtypes display less complex genetics than their high-grade counterparts harboring known specific,recurrent genetic alterations in the form of supernumerary ring chromosomes containing amplified material from 12q13-15 similar to that seen in some liposarcomas [Figure 8][83-87].In some cases,these changes have been identified in high-grade osteosarcoma as well,although it is unclear if this represents an isolated finding in a genetically complex lesion or a marker of transition from a previously low-grade lesion (dedifferentiation)[83].Amplification of material from 12q13-15 leads to the amplification of multiple genes involved in tumorigenesis includingMDM2,CDK4,andFRS2[83].These amplifications can be detected by immunohistochemistry for MDM2 and CDK4,although testing by FISH or chromosomal microarray provides a more sensitive test[83].These findings are quite helpful when dealing with a low-grade fibroblastic proliferation as these tumors can share morphologic overlap with other fibroblastic tumors of bone.

Figure 6.Representative example of chromothripsis:CMA plot of chromosome 12 demonstrating 14 alternating CNAs involving the proximal region of the short (p) and long (q) arms of chromosome 12.This pattern of alternating CNAs is suggestive of“chromothripsis”.In addition,there is a deletion of the remaining distal portion of 12p.Sample was tested using the Agilent 180k aCGH+SNP oligo array,and data analysis was performed using the Agilent CytoGenomics software v4.0 (Image courtesy of Dr.Fady M.Mikhail,MD PHD,University of Alabama at Birmingham).CMA:chromosomal microarray;CNAs:copy number abnormalities
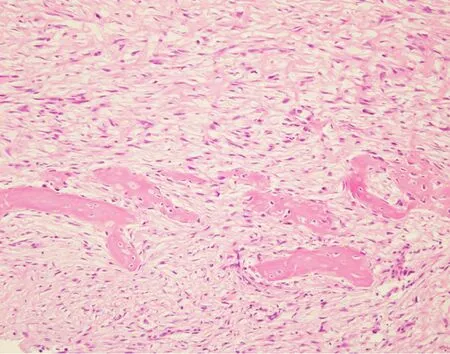
Figure 7.Parosteal osteosarcoma:high power magnification shows a bland appearing spindle cell population infiltrating around small bony fragments.Lesions such as this are difficult to distinguish from reactive processes or other fibroblastic lesions based on histomorphology alone (Hematoxylin and Eosin,20x magnification)
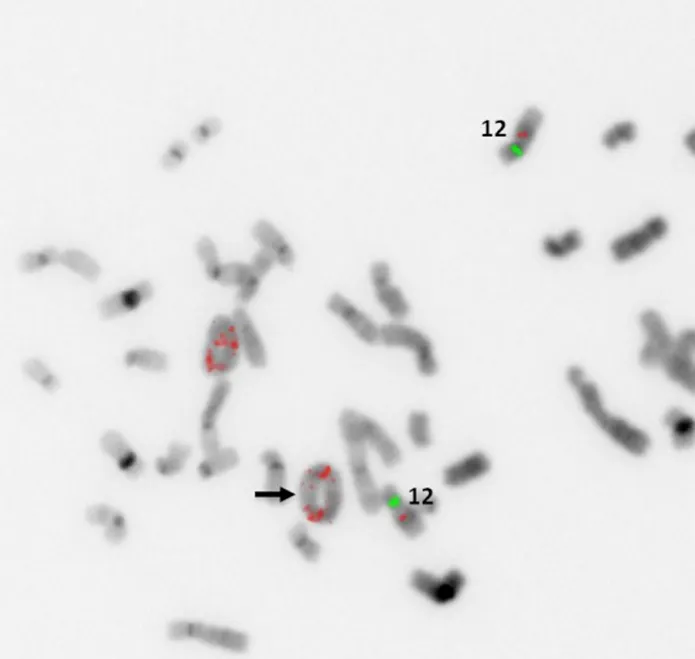
Figure 8.MDM2 amplification by FISH:representative example of supernumerary ring chromosome formation containing amplified material from 12q13-15.Increased copy numbers of MDM2 (red probe) signals (black arrow) are seen within ringed chromosomes(Image courtesy of Dr.Christine Bryke MD,Beth Israel Deaconess Medical Center).FISH:fluorescence in situ hybridization

Figure 9.Sclerosing epithelioid fibrosarcoma/low-grade fibromyxoid sarcoma:Scanning magnification of FUS/LGFMS showing an infiltrative,but bland appearing fibroblastic lesion showing both intramedullary and extramedullary growth (Hematoxylin and Eosin,20x magnification)
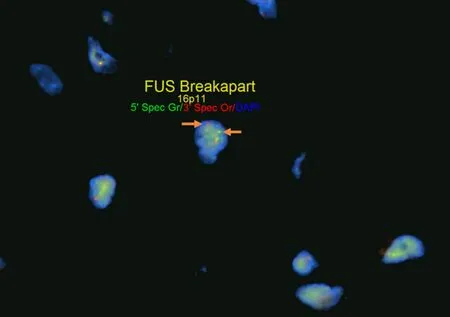
Figure 10.FUS fluorescence in situ hybridization (FISH):FISH for FUS in sclerosing epithelioid fibrosarcoma/low-grade fibromyxoid sarcoma using break apart FISH probes identifies a rearrangement of FUS indicated by separate signals (white arrows,5’ green probe and 3’ red probe).Testing for rearrangements through FISH does not provide information on partner genes or chromosomes involved in rearrangements
Ewing Sarcoma/Primitive Neuroectodermal Tumor
Ewing sarcoma is the prototypical example a small round blue cell sarcoma that may occur as primary bone tumor.Ewing sarcoma is characterized by a recurrent,specific t(11;22)(q24;q12) that leads to the production of an oncogenic fusion protein:EWSR1-FLI1[88].Various other fusion partners exist forEWSR1[Table 3],although many of these tumors are considered to fall within the Ewing sarcoma family and are treated in a similar fashion[89-92].

Table 5.Molecular genetic alterations in soft tissue tumors that may also rarely occur as primary bone tumors (simple and complex genetics)
The differential of small round blue cell sarcomas within bone has expanded considerably in the past years and now includes various non-Ewing translocation associated sarcomas,mesenchymal chondrosarcoma,myoepithelial tumors,and other poorly differentiated malignancies including high-grade osteosarcoma[90].The wide range of lesions that can share morphologic overlap makes molecular testing forEWSR1essential in the diagnosis of these lesions.While FISH is helpful in identifying the involvement ofEWSR1in a rearrangement,more advanced molecular techniques such as PCR or NGS may be required in order to identify fusion partners and determine the exact nature of a small round blue cell sarcoma.
SOFT TISSUE TUMORS THAT RARELY PRESENT AS PRIMARY BONE TUMORS
Apart from tumors that occur commonly within bone or primarily within bone there is a large group of mesenchymal neoplasms that may also rarely occur in bone.This list includes,but is not limited to,neural tumors (e.g.,malignant peripheral nerve sheath tumor),adipocytic tumors (e.g.,liposarcoma),histiocytic tumors (e.g.,Rosai-Dorfman disease),tumors of uncertain histogenesis (e.g.,synovial sarcoma),vascular tumors (e.g.,epithelioid hemangioma and angiosarcoma),and some undifferentiated sarcomas [Table 5][122-183].These lesions tend to share similar genetic profiles to their soft tissue counterparts when occurring as a bone primary and include lesions that fall into the simple and complex genetic category.Molecular diagnostic testing is indicated in many of these tumors as several of them are characterized by specific,recurrent alterations.An example of this is sclerosing epithelioid fibrosarcoma/low-grade fibromyxoid sarcoma - a bland appearing fibroblastic lesion that can share morphologic overlap with reactive changes,low-grade variants of fibroblastic osteosarcoma,desmoplastic fibroma,solitary fibrous tumor,and others [Figure 9].These tumors are characterized byFUSrearrangements,usually partnering withCREB3L2orCREB3L1and rarely occur within the bone; however,may pose problems for diagnosis when encountered[131-133].Although immunohistochemistry for MUC4 is helpful for the diagnosis,ancillary molecular testing through FISH,PCR,or sequencing can help solidify the diagnosis [Figure 10].While some of these lesions can be diagnosed based on clinicopathologic features and immunohistochemistry,oftentimes they require expanded diagnostic testing for specific molecular alterations.
CONCLUSION
Bone tumors represent a heterogenous category of benign and malignant lesions with a varied genomic landscape.Advances in molecular technology have vastly increased our knowledge of the molecular features of many of these lesions although the diagnosis of many bone tumors is still based entirely on histopathologic,clinical,and radiological features.Ancillary molecular diagnostics are increasingly becoming necessary for the diagnosis of bone tumors,facilitated by the development of new technologies in the past few decades.Knowledge of the molecular alterations as well as specimen handling considerations that may affect molecular testing is of utmost important as our base of knowledge continues to grow.
DECLARATIONS
Authors’ contributions
Responsible for conceptualization,image procurement,and content review: Suster S
Rresponsible for drafting the primary manuscript,image procurement,and figure creation: Suster D.
Availability of data and materials
Not Applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not Applicable
Consent for publication
Not Applicable
Copyright
© The Author(s) 2021.
