
缢蛏5个群体遗传多样性和遗传分化的SNP分析
2021-07-17 03:25滕爽爽胡高宇范建勋柴雪良肖国强
水生生物学报 2021年4期
滕爽爽 胡高宇 范建勋 柴雪良 肖国强
(1. 浙江省海洋水产养殖研究所, 浙江省近岸水域生物资源开发与保护重点实验室,温州市海洋生物遗传育种重点实验室, 温州 325005; 2. 三门县农业农村局, 台州 317100)
缢蛏(Sinonovacula constrictaLamarck), 属软体动物门(Mollusca), 瓣鳃纲(Lamellibranchia), 帘蛤目(Veneroida), 竹蛏科(Solenidae), 缢蛏属(Sinonovacula), 主要分布于日本、韩国、越南和我国的沿海、河口及中低潮间带区域, 为广温广盐海产双壳贝类。缢蛏生长快, 养殖周期短, 味道鲜美, 具有较高的市场价值, 据FAO[1]统计, 2016年我国缢蛏产量达82.3×107kg, 占蛏类总产量的95.4%, 是我国沿海滩涂贝类主要养殖品种之一。缢蛏养殖过去主要集中在浙江和福建一带, 近年来, 随着缢蛏消费市场的扩大及人工育苗技术的成熟, 缢蛏养殖业迅速发展, 山东沿海地区从浙江、福建引进苗种进行培育养殖; 浙江沿海为了能提早出苗, 每年9月从福建和广东等地购买亲贝, 催产育苗, 培育的苗种又销往山东、福建、广东和广西等地。频繁地异地引苗、盲目引种和无序养殖等可能使得缢蛏引入群体和当地群体发生基因交流, 导致缢蛏群体遗传多样性降低, 缢蛏当地种质资源混杂等问题。
目前, 关于缢蛏群体遗传结构和遗传多样性的报道主要采用同工酶、线粒体COⅠ、RAPD、AFLP、ISSR、ITS和SSR等标记来进行研究, 如王冬群等[2]利用同工酶分析野生群体和养殖群体的遗传多样性; Tran等[3]基于mtDNA-COⅠ和ITS2技术,研究了中国、韩国和越南缢蛏种群的遗传结构;Wang等[4]运用AFLP标记分析了缢蛏自然群体的遗传变异; 刘博等[5]利用微卫星标记技术研究了4个缢蛏群体的遗传多样性和系统发生关系等。单核苷酸多态性标记(SNP)是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性, 与上述标记相比, SNP标记具有以下优点: (1)数量多、分布广、遗传稳定和覆盖密度大; (2)某些SNP与生物特定性状直接相关; (3)SNP在群体中的分布频率具有显著差异, 更适合用于与性状的关联分析; (4)SNP的二态性特征使得其便于自动化检测。SNP标记已经广泛应用于群体遗传结构分析[6—10]、高密度遗传连锁图谱构建[11—15]、性状关联分析[16—20]、分子标记辅助育种[21,22]和亲权鉴定[23]等方面, 并表现出良好的应用前景。SNP标记应用于水产动物群体遗传结构分析已经有若干报道, 如日本鳗鲡[24]、大黄鱼(Larimichthys crocea)[25]、马氏珠母贝(Pinctada fucata)[26]和栉孔扇贝(Chlamys farreri)[27]等。
本研究将采用多态性SNP分子标记对山东、浙江、福建、广东和广西等地缢蛏群体的遗传结构进行分析, 以期进一步揭示我国沿海缢蛏理群体的遗传背景, 探讨缢蛏群体遗传多样性和遗传分化水平, 分析缢蛏地理群体遗传变异与地理分布的关系, 为缢蛏地理群体的识别、种质资源保护及良种选育提供基础资料。
1 材料与方法
1.1 样品采集
2017年9—10月, 缢蛏5个种群分别采自山东东营(DY)、浙江乐清(YQ)、福建云霄(YX)、广东湛江(ZJ)和广西钦州(QZ)等地(图1)。缢蛏山东东营种群采自东营河口区, 是滩涂上自然繁殖自然生长群体; 其他4个群体苗种分别来源于当地自然产苗区, 人工采苗, 池塘养殖。每个群体取40个样品, 解剖样品, 取足于70%酒精中保存。样品信息见表1。

图1 缢蛏群体样品采集点Fig. 1 Sample location sites of S. constricta populations

表1 缢蛏5个种群的样品信息Tab. 1 Sample information of S. constricta populations
1.2 DNA提取、PCR扩增及产物检测
DNA提取基因组DNA采用艾德莱组织/细胞基因组DNA快速提取试剂盒(DN0711)进行抽提,提取出来的DNA, 用1%琼脂糖电泳检测DNA的完整性, Nano-400超微量核酸分析仪检测DNA纯度和浓度, 再配制成10 ng/μL工作液, 4℃备用。
SNP位点序列信息获得本研究所采用的缢蛏SNP位点来源于已发表文献[28], 参考文献中18个位点的引物序列及PCR反应体系和反应程序,随机扩增18个个体, 双向测序和拼接, 得到位点所在的序列信息。采用Primer-blast方法设计SnaPshot扩增引物和延伸引物(表2)。
SNaPShot方法检测样品首先, 采用扩增引物获得PCR产物并纯化。PCR反应体系为20 μL,包括KAPA 2G Robust HotStart ReadyMix(Roche)17.0 μL, 10 mmol/L扩增引物各1.0 μL, DNA模板1.0 μL。PCR反应程序为: 98℃变性2min, 进入35个循环, 每一循环包括98℃变性10s, 复性(Tm温度见表2)10s, 72℃延伸10s, 最后一步是72℃延伸5min。最后将产物于4℃下保存。扩增产物用1.5%琼脂糖凝胶检测, 鉴定有扩增产物后, 以快速PCR产物磁珠法回收纯化试剂盒(志昂, GO-PCRF-5000)纯化PCR产物。
其次, 延伸标记和纯化。SnaPshot PCR反应体系为5 μL, 包括SNaPshot Multiplex Ready Reaction Mix(ABI)0.8 μL, 模板(上述纯化PCR产物)2.0 μL,延伸引物(表2)1.2 μL, 加ddH2O至5 μL。PCR反应程序为: 96℃变性5min, 进入25个循环, 每一循环包括96℃变性10s, 复性(Tm温度见表2)5s, 60℃延伸30s, 最后一步是60℃延伸5min。在反应结束后, 进行第2次纯化, 在每个SnaPshot PCR产物中, 加入SAP(1 U/μL)0.5 μL, 10×SAP Buffer 0.5 μL, 37℃孵育1h, 75℃ 15min灭活SAP酶, –20℃保存。

表2 缢蛏SNaPshot扩增引物信息Tab. 2 Sequences of SNaPshot amplification primers of S. constricta
最后, 检测SNP位点基因型。反应体系5 μL, 包括模板(第2次纯化产物)0.8 μL, HiDi+GS120 (ABI)预混液4.2 μL。95℃变性5min, 待温度降到4℃后拿出, 短离心1000 r/min后, 使用ABI 3730进行毛细管电泳, GeneMapper 4.1 进行分型统计。
1.3 数据分析
根据缢蛏5个群体15个SNP位点基因型统计结果, 应用Popgene 1.32 软件计算每个SNP位点的有效等位基因数(Ne)、shannon指数(I)、期望杂合度(He)、观察杂合度(Ho)、最小等位基因频率(MAF)、哈代-温伯格平衡值(Hardy-Weinberg equilibrium,HWE)、Nei’s遗传相似度和遗传距离。通过STRUCTURE V2.3.3[29]判断5个群体200个个体的遗传结构, 根据DeltaK值确定最佳类群数, 对于确定的K值, 重复运行Structure程序1000次, 每次在10000burn-in基础上运行10000步, 获得群体的遗传结构图由Distruct程序构建。采用Arlequin3.5软件对群体进行AMOVA(Analysis of molecular variance)分析, 并计算群体间遗传分化(Fst)。根据群体间遗传距离数据, 利用MEGA4.0对5个群体进行聚类分析, 构建UPGMA聚类树, 以此来确定各个群体的亲缘关系远近。
2 结果
2.1 SNaPshot扩增引物信息获得
位点ScSNP-5、ScSNP-11和ScSNP-16测序结果存在信号衰减和双峰现象, 因此在后续实验中去掉这3个位点。其余15个位点各扩增18个个体, 经双向测序和拼接后获得序列信息。根据SNaPshot引物设计原则, 参考SNP位点来源文献[28]中各位点的突变碱基, 15个位点均各获得2条扩增引物序列和1条延伸引物序列(表2)。
2.2 缢蛏群体遗传多样性分析
对15个SNP位点在5个群体的分型结果进行遗传结构统计分析, 结果显示, DY、YQ、YX、ZJ和QZ的平均有效等位基因数(Ne)分别是1.754、1.555、1.558、1.533和1.519; 平均香农指数(I)为0.605、0.501、0.502、0.489和0.471; 平均观测杂合度(Ho)为0.317、0.282、0.282、0.265和0.285; 平均期望杂合度(He)为0.423、0.336、0.338、0.325和0.313; 平均最小等位基因频率(MAF)为0.336、0.241、0.238、0.234和0.229(表3); 高多态性SNP(MAF≥0.30)位点数为9、5、5、4和4(图2)。

图2 缢蛏群体15个SNP位点最小等位基因分布Fig. 2 Minor allele distribution across five S. constricta populations using 15 single nucleotige polymorphisms

表3 基于15个多态性SNP位点的缢蛏群体遗传多样性分析Tab. 3 Genetic diversity summary in five populations of S. constricta from different SNPs
2.3 缢蛏群体结构分析
采用STRUCTURE软件分析群体结构, 假设类群值K在3—10, 进行聚类。根据DeltaK值最大来确定最佳分类群数是5(图3), 表明200个缢蛏个体可以划分为5个类群。基于15个SNP标记分析各个样品的遗传组分在各个聚类簇中的百分比, 以75%类群属性比作为划分依据, 200个个体类群归属结果如图4。在5个聚类簇中, 聚类簇1、聚类簇2、聚类簇3、聚类簇4和聚类簇5分别包含45、38、46、38和33个个体。表4显示了5个群体在每个聚类簇中所占比例, 东营群体中有39.7%的个体归属到聚类簇5, 归属到聚类簇1—4的个体在11.9%—17.4%,乐清群体和云霄群体分别有34.9%和34.6%的个体归属到聚类簇3, 湛江群体中有31.3%的个体归属到聚类簇1, 钦州群体中有30.8%的个体归属到聚类簇2。
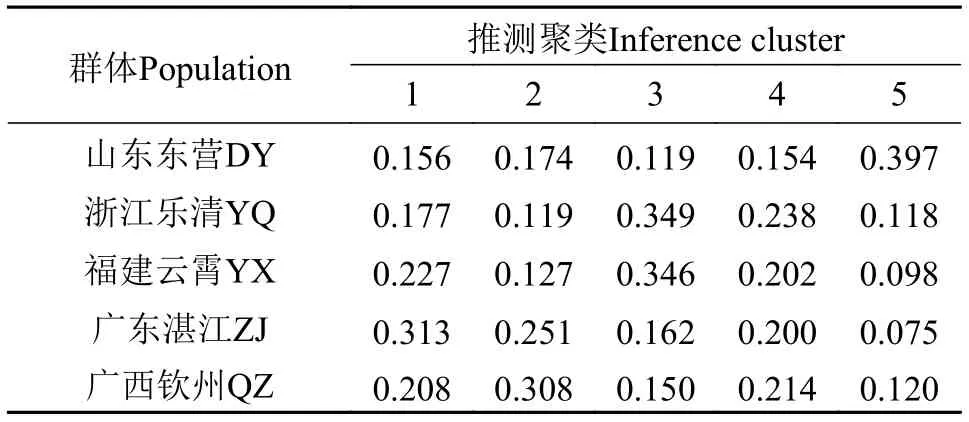
表4 缢蛏群体在5个聚类簇占比分析Tab. 4 Proportion of analyzed S. constricta populations in each of the five clusters

图3 LnP(D)值随K值的变化曲线Fig. 3 The LnP (D) statistic for each given K

图4 基于模型的群体结构分析Fig. 4 Model-based ancestries
2.4 缢蛏群体AMOVA分析及遗传分化
应用AMOVA对缢蛏5个群体的遗传变异进行分析(表5), 根据养成方式, 5个地理群体分为2组,即野生群体组和养殖群体组, 来自组间、群体间和群体内个体间的遗传变异分别为4.67%, 3.31%和92.02%(P<0.05)。按照地理分布, 分为3组, 93.38%的变异来自群体内个体间(P<0.05)。缢蛏群体两两之间的遗传分化(Fst)值在–0.0061—0.0829, 其中乐清群体和云霄群体, 湛江群体和钦州群体Fst无显著差异(P≥0.05), 其余群体两两之间Fst差异显著。Reynolds’遗传距离在0.000(湛江群体和钦州群体)–0.0865(东营群体和湛江群体)之间(表6)。

表5 缢蛏5个群体的AMOVA分析Tab. 5 Analysis of molecular variance among S. constricta populations

表6 遗传分化和Reynolds’遗传距离Tab. 6 Pairwise genetic differentiation (Fst) and Reynolds’ genetic distance among five S. constricta populations
2.5 缢蛏群体遗传距离及聚类分析
基于15个SNP位点分析缢蛏5个地理群体的Nei’s遗传相似度和遗传距离, 结果如表7。群体两两之间遗传距离在0.0023—0.0537, 遗传相似度在0.9477—0.9977, 群体间较高的遗传相似度说明5个地理群体具有相似的遗传结构。其中, 湛江群体和钦州体遗传距离最小(0.0023), 乐清群体和云霄群体遗传距离次之(0.0026), 东营群体与湛江群体遗传距离最远(0.0537)。Nei’s遗传聚类结果显示, 乐清群体在云霄群体聚在一起, 湛江群体和钦州群体聚在一起, 东营群体与其他4个群体遗传距离最远,单独为一支(图5)。
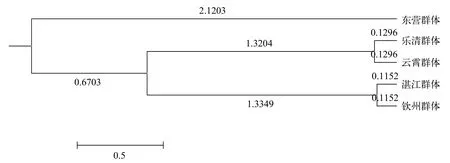
图5 基于缢蛏群体间遗传距离构建的聚类图Fig. 5 UPGMA clustering of samples based on Nei’s genetic distance

表7 Nei’s遗传相似度和遗传距离Tab. 7 Nei’s genetic identity and genetic distance
3 讨论
3.1 缢蛏群体遗传多样性分析
群体遗传学分析指标中, 遗传参数(有效等位基因数(Ne)、香农指数(I)、观测杂合度(Ho)、期望杂合度(He)和最小等位基因频率(MAF)等的大小反映了群体遗传多样性水平, 其数值越大, 说明基因丰富度越高。本研究的结果表明, 5个群体的Ne为1.519—1.754,I为0.471—0.605,Ho为0.265—0.317,He为0.313—0.423, MAF为0.229—0.336。Jiang等[25]利用飞行质谱阵列SNP基因分型技术(Sequenom MassARRAY)分析了大黄鱼野生群体的遗传多样性,Ne为1.328—1.341,Ho为0.190—0.253,He为0.273—0.320; Dong等[30]的利用高分辨率溶解曲线法(High-resolution melting, HRM)分析了海参野生群体的遗传多样性,Ho为0.2664—0.2760,He为0.3188—0.3280。缢蛏群体的杂合度值高于大黄鱼和海参, 因此, 我们推测5个缢蛏群体的遗传多样性处于较高水平, 具有较高的遗传变异潜力。与乐清群体、云霄群体、湛江群体和钦州群体等4个群体相比, 东营群体的遗传参数值更大, 表明其遗传多样性更高。遗传多样性降低, 对经济性状, 如存活率、生长率和疾病耐受性等可能产生不利的影响,而更高水平的遗传多样性, 更加广泛的变异, 有利于生物群体适应环境的变化, 维持长期的生存, 因此, 有必要使用分子标记评估和监控缢蛏群体的遗传多样性。
Niu等[31]利用SSR标记分析缢蛏群体遗传结构,Ho为0.737—0.909,He为0.836—0.882; 刘达博等[32]同样采用SSR分析了乐清湾和三沙湾缢蛏群体遗传多样性, 其He分别为0.872—0.909和0.846—0.894, 均高于本研究的缢蛏群体的Ho和He值。在变异度丰富的群体中, 每个微卫星位点可以有更多数量的基因型, 而每个SNP位点最多只有3种基因型, 因此, 在群体遗传学研究中需要使用更多数量的SNP位点。但是, 对于SNP标记来说, 由于其等位基因数量少, 使得分型快速, 错误率更低, 并且SNP作为基因组中最丰富的变异的特征增加了其多态性[33]。
3.2 缢蛏群体遗传分化
Wright[34]建议群体间遗传分化Fst值在0—0.05,表明群体间遗传分化很小, 可以不考虑;Fst值在0.05—0.15, 为中等程度分化;Fst值在0.15—0.25, 为较大程度分化;Fst值在0.25以上, 则群体间有很大的遗传分化。乐清群体和云霄群体、乐清群体和钦州群体、云霄群体和钦州群体、湛江群体和钦州群体之间Fst值为–0.0061—0.0446, 遗传分化很小, 其余两两群体之间为中等程度分化。AMOVA分析结果显示90%以上的遗传变异来自群体内, 也说明了群体间没有明显的遗传分化。Thorp[35]认为独立群体间的Nei’s遗传距离在0.030—0.200。乐清群体和云霄群体、乐清群体和钦州群体, 云霄群体和钦州群体, 湛江群体和钦州群体之间的Nei’s遗传距离都小于0.030, 说明这几个群体两两之间不是完全独立的群体。利用STRUCTURE软件对缢蛏200个个体进行群体结构分析,K值变化曲线结果表明缢蛏200个个体可以划分为5个聚类簇。每个群体的40个个体在5个聚类簇中都有分布, 分布数量在3至16个, 分布比例均没有超过0.75, 即每个群体没有显著的聚集到某一个聚类簇中, 说明每个群体不同个体之间存在较大的遗传差异。5个群体在同一聚类簇中的分布比例都相似。这两点与AMOVA分析得出的结果一致, 即遗传变异主要存在与群体内。
配子和幼虫扩散是海洋底栖无脊椎动物在自然条件下发生基因流的主要原因[36]。乐清群体和云霄群体这2个采样地之间直线相距约为588 km,缢蛏幼虫在变态附着前, 平均浮游期为4—7d, 不足以达到有效的扩散。缢蛏钦州群体与湛江群体在地理位置上比较近(直线距离约220 km), 而雷州半岛却将两个地区分隔开, 且雷州半岛对海流有阻隔作用, 因此自然水流不大可能是浮游幼虫扩散的原因。另一方面不同的气候导致了不同的繁殖时间,广东和广西缢蛏的性腺成熟时间较早, 主要集中在8月初; 越往北, 性成熟时间越晚, 浙江通常在10月初。这也使得不同地区的缢蛏群体之间存在生殖隔离和有限的基因流。
乐清群体和云霄群体、钦州群体和其余4个群体间的Fst值和Nei’s遗传距离揭示出这几个群体之间违反距离隔离模型, 存在非典型模式。缢蛏养殖过去主要集中在浙江和福建一带, 浙江育苗企业从福建采购亲贝进行人工育苗, 在苗种繁育过程中,有限的亲本数量降低了有效群体大小, 培育的苗种又销往福建; 同时, 浙江也从福建采购苗种进行中培和养殖。自2012年以来, 浙江和福建等地育苗企业为了提早出苗, 每年8—9月从广西等地采购亲贝进行人工繁殖, 培育出的苗种一部分在本地进行中培和养殖, 其余大部分销往辽宁、天津、山东、福建、广东和广西等地。因此, 本研究中缢蛏群体的苗种来源虽为当地自然苗种, 但是由于频繁的引种,苗种的迁移, 人工培育苗种与当地自然苗种存在基因交流, 可能对缢蛏基因型的产生了影响, 干扰和混杂了缢蛏当地种质资源, 这可能是导致缢蛏乐清群体和云霄群体, 钦州群体和其他群体基因交流频繁的, 群体内遗传变异大, 群体间遗传分化不高的主要原因。在缢蛏群体遗传结构和遗传分化研究中, 我们发现了类似的结果, 刘博等[27]对缢蛏4个养殖群体进行聚类分析, 发现遗传距离和地理距离并未表现出正相关, 认为这可能与缢蛏无序移养有关。Niu等[30]对沿海10个群体SSR分析中, 发现缢蛏北方群体(青岛和海阳)和南方群体(宁波、台州、宁德和漳州)聚为一支, 推测原因是由于北方引入浙江和福建的苗种, 导致南北群体间存在基因流。
3.3 缢蛏种质资源的保护与开发
本研究采用15个多态性SNP标记研究缢蛏群体遗传结构, 发现缢蛏5个群体遗传多样性丰富, 而群体间遗传分化程度不高, 地理群体之间违反距离隔离模型。在缢蛏群体遗传学已有的研究中, 我们发现在2012年之前的研究中, 如Wang等[10]采用AFLP标记研究辽宁至广西6个缢蛏群体的遗传结构, 发现群体遗传多样性高, UPGMA聚类分析结果显示群体间遗传距离与地理距离符合。而在2012年之后的研究中, 如Niu等[30]采用8个微卫星标记分析中国南北沿海10个缢蛏群体的遗传分化, 刘博等[27]采用12个微卫星标记分析缢蛏4个养殖群体的系统发生关系, 都发现南北方群体之间存在基因交流, 主要的遗传变异来自群体内, 这与2012年之后, 浙江和福建等地出现亲贝引种和苗种移养现象有极大关系, 人为干扰可能是导致这种情况出现的主要原因。缢蛏是我国主要养殖滩涂贝类之一, 加强当地缢蛏种质资源保护, 一方面, 可以建立原、良种场和自然保护区; 另一方面, 在育苗和养殖方面, 要合理有序的引种, 减少或防止与当地群体混杂。
猜你喜欢
水运工程(2022年10期)2022-11-02
当代水产(2021年11期)2022-01-15
水上消防(2021年4期)2021-11-05
科学养鱼(2021年4期)2021-05-19
模具制造(2019年4期)2019-12-29
当代水产(2019年9期)2019-10-08
传媒评论(2019年4期)2019-07-13
中国水产(2018年10期)2018-10-24
陶瓷科学与艺术(2018年1期)2018-07-13
传媒评论(2018年3期)2018-06-21
