
醋酸反应釜液中各组分的测定
2021-07-07 06:06:18尹永志
河南化工 2021年6期
尹永志
(河南龙宇煤化工有限公司,河南 永城 476600)
公司醋酸装置设计年产能力40万t,利用一氧化碳和甲醇在反应器内进行低压羰基化反应合成醋酸,反应器内生成的醋酸和反应液以混合物的形态经降压闪蒸进一步精馏提纯,可产出99.9%的产品醋酸。反应釜内组分复杂,色谱和滴定得出的数据会忽略含碘物质在反应液中所占比例的影响,故采用气相色谱修正面积归一法测定水、碘甲烷、乙酸甲酯、乙酸的含量,硝酸银滴定法测定碘离子含量,通过含碘物质在反应液中所占比例,折算得出其他组分的准确含量,对工艺操作有很好的指导作用[1-2]。
1 气相色谱实验部分
1.1 方法原理
采用气相色谱修正面积归一法分析样品中水、碘甲烷、乙酸甲酯、乙酸含量,利用校正峰面积定量,保留时间定性[3]。
1.2 仪器与实验条件
载气,N2(99.99%以上),流速20 mL/min;辽宁科瑞GS-2010气相色谱仪;色谱柱,Porapak T;检测器,热导检测器(TCD),桥电流100 mA;进样器,170.0 ℃;柱箱,130.0 ℃;程序升温,4 min后以20.0 ℃/min的速度升温至200.0 ℃,保持15 min。
1.3 标准样品的配制
采用称重法配制与样品组分浓度相近的标准溶液,准确至0.000 1 g。于100 mL干燥的容量瓶中准确称取水7.115 0 g,碘甲烷20.206 2 g,乙酸甲酯0.982 5 g,乙酸79.691 1 g,混匀。通过计算得出各组分质量分数分别是:水6.59 %,碘甲烷18.71 %,乙酸甲酯0.91 %,乙酸73.79 %。碘甲烷易挥发,所以标准样品需现配现用(所用试剂均用卡尔费休法测定其中水的含量,消除试剂水的影响)。
1.4 标准曲线的建立
开启载气,打开色谱电源,调用按上述参数设置好的分析方法,待基线平直,仪器状态稳定后,用10 μL进样针吸取1 μL标准样品,快速插入进样口,按开始键运行,重复进样3~5次,选择合适的谱图做校正曲线,并存为模板。校正表见表1。
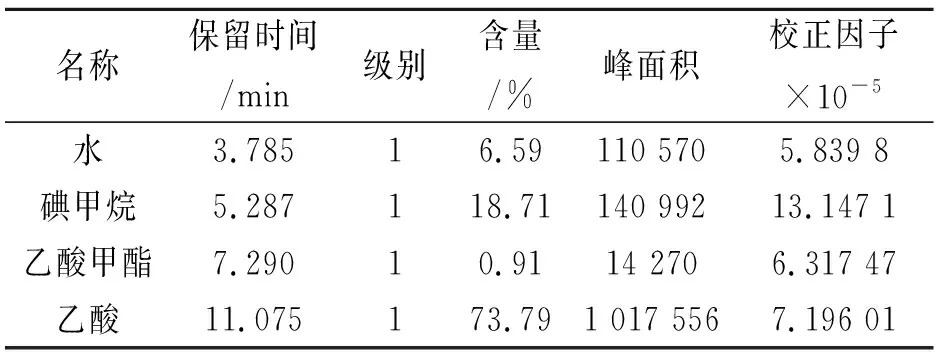
表1 校正表
1.5 样品的测定
按上述仪器条件和操作方法,混匀样品后用进样针吸取1 μL反应釜液样品进行分析,调用模板后,在定量结果界面点击定量计算,查看分析结果。

表2 试样结果
2 沉淀滴定分析碘离子
2.1 方法原理
反应液中还含有一定量的助催化剂氢碘酸、碘化锂等,而上述色谱方法无法分析其含量,固采用硝酸银沉淀滴定法测定。

2.2 仪器和试剂
酸式滴定管,0.1 mol/L硝酸银标准溶液。0.2%的曙红Y指示剂:0.2 g曙红Y溶于100 mL甲醇中。碳酸铵溶液:2.4 g碳酸铵用高纯水稀释到100 mL(现配现用)。
2.3 分析步骤
用天平准确称取0.5 g样品于盛有50 mL高纯水的锥形瓶中,加入5 mL乙酸,5 mL碳酸铵和5滴曙红Y指示剂,用0.1 mol/L的硝酸银滴定至出现亮红色,同时可见渣样沉淀即为终点[4]。
2.4 I-含量的计算
式中:V,滴定消耗的硝酸银标液体积,mL;c,硝酸银标液的浓度,mol/L;W,样品的质量,g。
3 结果的计算
由于修正面积归一法未能分析出I-含量,故其他组分含量需按百分比再进行折算。
即:ω水=A×(1-I-ωI-)×100%,A,色谱显示水的百分含量;ω碘甲烷=B×(1-I-ωI-)×100%,B,色谱显示碘甲烷的百分含量;ω乙酸甲酯=C×(1-I-ωI-)×100%,C,色谱显示乙酸甲酯的百分含量;ω乙酸=(1-ω水-ω碘甲烷-ω乙酸甲酯-ωI-)×100%。
4 精密度与准确度
4.1 精密度
同一个反应釜液样品充分混匀后,按上述条件连续分析3次,进行精密度验证,具体数据见表3。

表3 精密度分析
4.2 准确度
由于修正面积归一不适于做回收率实验,仅对滴定法做回收率实验。

表4 回收率实验
5 结论
经过精密度和准确度的验证,无论是色谱出峰时间的稳定性,还是滴定的精密度和准确度都能满足实验要求,而且色谱法操作简单,重现性好,对进样量要求不严格。通过折算后计算的组分含量更加准确,是测定醋酸反应液组分含量的一种不错选择。但是,在测定过程中也应注意以下问题: ①在配制标准样品时,要先用卡尔费休计算出所用试剂的水分含量,剔除试剂水的影响,由于碘甲烷挥发性大,所以为保证准确性,所配标样要现配现用。②反应液中含有大量金属离子,长期使用后衬管变脏,要定期清理衬管。③色谱进样前要混匀样品。
猜你喜欢
云南化工(2021年11期)2022-01-12 06:06:20
环境卫生工程(2021年5期)2021-11-20 05:45:30
疯狂英语·新悦读(2021年5期)2021-06-08 01:54:10
云南化工(2020年11期)2021-01-14 00:50:52
广州化工(2020年8期)2020-05-19 06:23:56
船电技术(2016年8期)2016-10-27 07:44:57
化工进展(2015年3期)2015-11-11 09:07:41
中国当代医药(2015年10期)2015-03-01 02:02:39
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:56
海军医学杂志(2015年2期)2015-02-27 13:47:35
