
6-硝基-4-芳氨基喹啉化合物的合成及体外抗肿瘤活性研究
2021-07-07 10:54薛艾奇汪海峰张昕旸
沈阳化工大学学报 2021年1期
刘 丹,薛艾奇,李 雪,栾 天,汪海峰,张昕旸
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
酪氨酸激酶表皮生长因子受体(EGFR)及其家族成员HER2是靶向癌症治疗的公认靶点[1].自20世纪80年代以来,当EGFR和HER2被提出作为潜在的抗癌靶点时,小分子激酶抑制剂已成为抑制EGFR和HER2激酶活性最具潜力的手段.迄今为止在美国食品和药物管理局(food and drug administration,FDA)批准用于靶向癌症治疗的抑制剂中,拉帕替尼(lapatinib,A)和埃罗替尼(erlotinib,B)属于4-苯氨基喹唑啉结构(见图1),治疗非小细胞肺癌或乳腺癌中发生的EGFR/HER2依赖性肿瘤[2-3].Pawar等[4]在4-苯氨基喹唑啉化合物的研究基础上合成一系列6-取代-4-苯氨基喹啉衍生物(C,见图1),证明以喹啉为母核的化合物同样具有较好的细胞毒活性;大量学者[5-8]在喹啉环的6位适当引入吸电子基,提高了化合物对HER家族的多重抑制作用,进而避免了耐药性的产生.课题组曾设计并合成了一系列7-氟-4-苯氨基喹啉化合物[9],部分化合物表现出较好的抗肿瘤活性.
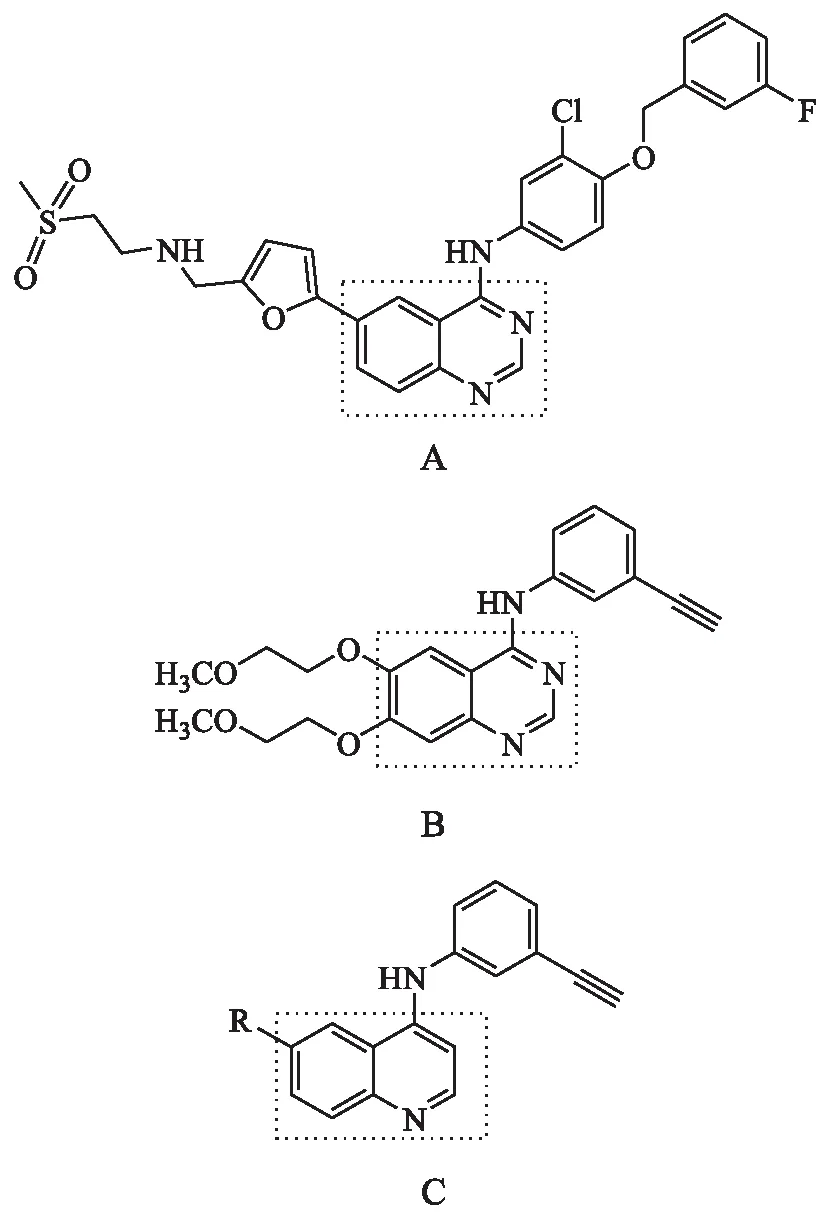
图1 酪氨酸激酶抑制剂Fig.1 Tyrosine kinase inhibitors
本文参考以上喹唑啉及喹啉化合物的构效关系,以喹啉为母核,在喹啉的6位连接硝基,4位上引入芳氨基,共合成4个6-硝基-4-芳氨基喹啉化合物(6a~6d),并以SGC-7901、BEL-7402和A549细胞为靶细胞,测定其体外抗肿瘤活性.合成路线及目标化合物结构如图2所示.
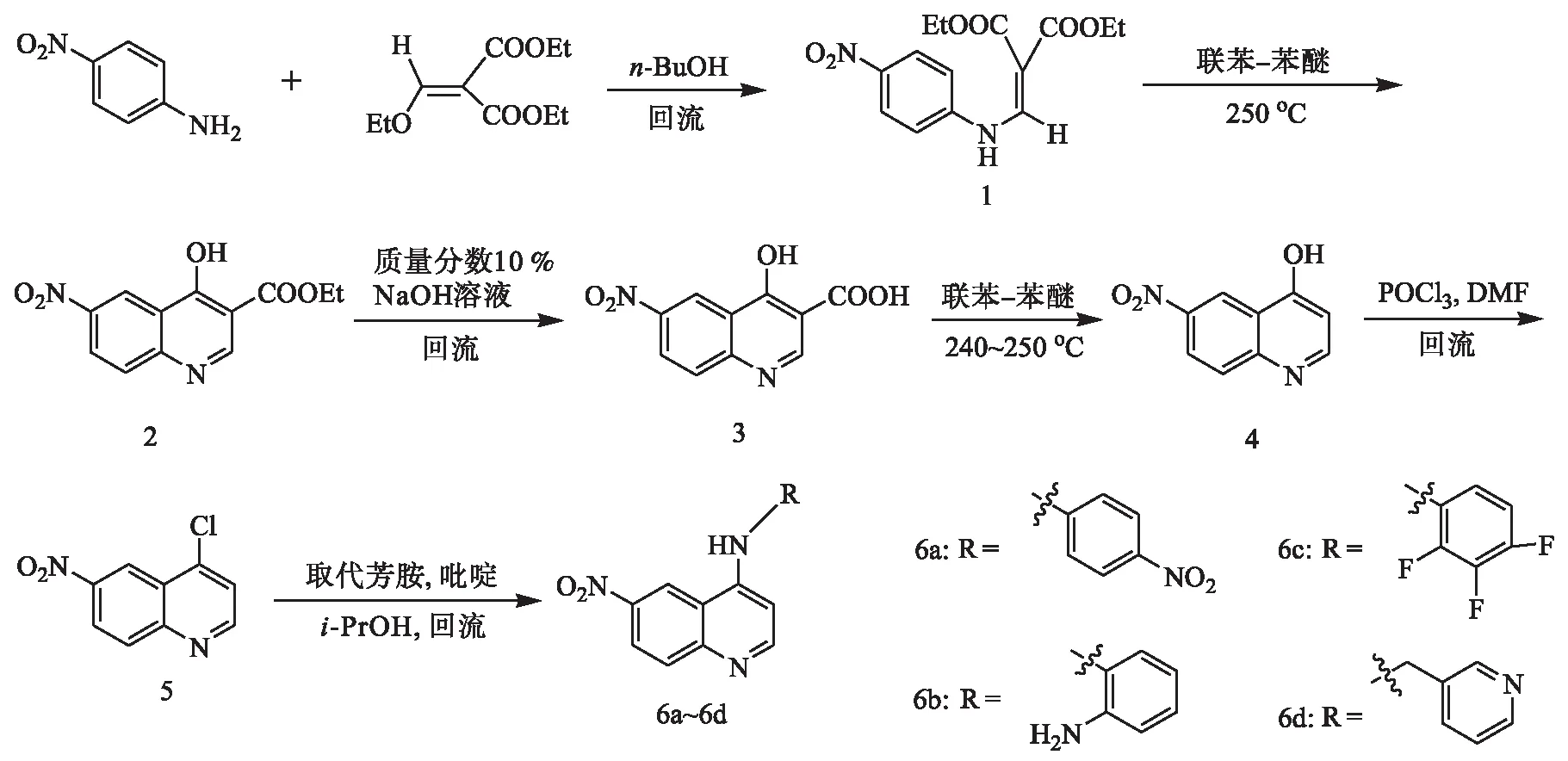
图2 目标化合物的合成路线Fig.2 Synthetic route of target compounds
1 实验部分
1.1 仪器与试剂
X-4型数字显微熔点测定仪,北京泰克仪器有限公司;ZF-7三用紫外分析仪,上海顾村电光仪器;Thermo-Finnigan LCQ型质谱仪,美国热电-菲尼根公司.
吉非替尼(gefitinib,质量分数99 %),齐鲁(海南)制药公司;RPMI1640 DMEM 培养基及胰酶,Gibeco公司;胎牛血清,北京鼎国生药技术有限责任公司;溴化四氮唑盐、蛋酶K和小牛血清蛋白,Sigma 公司;SGC-7901细胞、BEL-7402细胞和A549细胞,中科院上海细胞库;其余所用试剂均为分析纯或化学纯.
1.2 合成
1.2.1 2-((4-硝基苯氨基)亚甲基)丙二酸二乙酯(1)的合成
取5.64 g(26.07 mmol)EMME与3.00 g(21.73 mmol)4-硝基苯胺置于三颈瓶中加入30 mL的正丁醇,回流搅拌反应6 h,反应结束.将剩余物中加入无水乙醇,冷却结晶,过滤,干燥后得5.82 g淡黄色针状固体1,收率86.92 %,mp 141.91~142.23 ℃.
1.2.2 4-羟基-6-硝基喹啉-3-羧酸乙酯(2)的合成
将40 mL的联苯-苯醚(质量比为1∶3)加入100 mL的三颈瓶中,磁力搅拌下加热至250 ℃,将2.00 g(6.51 mmol)化合物1分批并缓慢加入联苯-苯醚中,250 ℃下反应1 h.冷却至室温,加入40 mL石油醚,析出固体.过滤,滤饼用石油醚洗涤,干燥得1.32 g白色固体2,收率76.63 %,mp 352.54~352.81 ℃.
1.2.3 4-羟基-6-硝基喹啉-3-乙酸(3)的合成
在100 mL三颈瓶中加入1.02 g(3.90 mmol)的化合物2与质量分数10 %的氢氧化钠溶液30 mL,搅拌下回流反应4 h.冷却至室温,用盐酸调pH至1,析出白色固体,抽滤,滤饼用水洗至中性,干燥后得0.85 g白色固体3,收率92.72 %,mp 284.54~286.43 ℃.
1.2.4 4-羟基-6-硝基喹啉(4)的合成
三颈瓶中加入0.50 g(2.17 mmol)化合物3和50 mL联苯-苯醚试剂,240~250 ℃下反应1 h,冷却至室温,加入50 mL石油醚,析出固体.抽滤,滤饼用石油醚洗涤,得0.32 g黄色固体4,收率91.44 %,mp 323.73~325.24 ℃.
1.2.5 4-氯-6-硝基喹啉(5)的合成
将0.30 g(1.58 mmol)化合物4溶于5 mL 三氯氧磷中,滴加1滴N,N-二甲基甲酰胺(DMF)后,于80 ℃下反应3 h,冷却至室温,将反应液缓慢倾倒在冰水中并不断搅拌,用饱和NaHCO3溶液调节pH至3,抽滤,滤饼水洗至中性,得白色固体,硅胶柱色谱纯化,洗脱剂为石油醚/乙酸乙酯(体积比为5∶1),得0.28 g白色固体5,收率85.42 %,mp 142.21~142.84 ℃(文献值[8]mp 144~145 ℃).
1.2.6 6-硝基-4-(4′-硝基苯氨基)喹啉(6a)的合成
将100 mg(0.48 mmol)化合物5,73.12 mg(0.53 mmol)4-硝基苯胺,38.00 mg(0.48 mmol)吡啶溶于30 mL异丙醇中回流,反应结束.减压蒸除异丙醇,冷却至室温,加入10 mL乙醚、10 mL饱和NaHCO3溶液,室温搅拌0.5 h,抽滤,滤饼用水洗至中性,硅胶柱色谱纯化,洗脱剂为石油醚/乙酸乙酯(体积比为5∶1),得117.10 mg黄色固体6a,收率78.53 %,mp 219.91~220.53 ℃,MS(ESI),m/z:311.82{[M+H]+}.
1.2.7 6-硝基-4-(2′-氨基苯氨基)喹啉(6b)的合成
化合物6b是由化合物5与2-氨基苯胺所得,制备方法如同化合物6a.黄色固体,收率79.13 %,mp 234.32~235.71 ℃,MS(ESI),m/z:281.73{[M+H]+}.
1.2.8 6-硝基-4-(2′,3′,4′-三氟苯氨基)喹啉(6c)的合成
化合物6c是由化合物5与2,3,4-三氟苯胺所得,制备方法如同化合物6a.黄色固体,收率82.64 %,mp 210.52~211.74 ℃,MS(ESI),m/z:320.71{[M+H]+}.
1.2.9 6-硝基-4-(吡啶-3-亚甲基苯氨基)喹啉(6d)的合成
化合物6d是由化合物5与3-氨甲基吡啶所得,制备方法如同化合物6a,但不加乙醚.黄色固体,收率81.52 %,mp 198.94~200.22 ℃,MS(ESI),m/z:281.14{[M+H]+}.
1.3 体外抗肿瘤活性筛选
采用MTT法检测4个6-硝基-4-芳氨基喹啉化合物对人胃癌细胞SGC-7901、人肝癌细胞BEL-7402和非小细胞肺癌细胞A549的体外抗肿瘤活性,选用吉非替尼作为阳性对照[10-11].3种细胞株均培养于含有10 %(体积分数)胎牛血清的RPMI 1640培养基中.用胰酶使细胞脱壁,以每孔5×103个细胞的密度种植在96孔板中,置于含5 %(体积分数)CO2的37 ℃恒温箱中培养过夜.然后将不同质量浓度(0.4 mg/L,1.2 mg/L,3.6 mg/L,11 mg/L,33 mg/L)的待测目标化合物以每孔20 μL的量加到96孔板中,并设置3个复孔,在恒温箱中孵育48 h.之后在每孔中加入新配置的MTT溶液,在37 ℃恒温箱中继续孵育4 h.每孔加入100 μL的DMSO使甲瓒完全溶解,用酶标仪在490 nm处测定各孔光密度(OD)值,计算所测目标化合物对肿瘤细胞的抑制率.IC50为48 h后抑制细胞数量为50 %的药物浓度.
2 结果与讨论
2.1 合成
在合成化合物1时,最初选用乙醇作为溶剂[12],由于硝基有强吸电诱导效应和共轭效应,使得硝基苯胺的氨基N原子的亲核能力降低,因此,选用沸点较高的正丁醇代替乙醇作为溶剂,回流反应,升高温度有利于反应的进行,与乙醇作溶剂相比反应时间大大缩短,通过TLC监测完全反应只需6 h.
2.2 体外抗肿瘤活性
体外抗肿瘤活性筛选结果见表1.由表1可知:化合物6a(喹啉环4位苯氨基上连接硝基)对SGC-7901和BEL-7402细胞无活性,但对A549有较强的抑制活性(IC50为9.32 μmol/L),且活性优于吉非替尼(IC50为 14.77 μmol/L);化合物6b(喹啉环4位苯氨基上连接氨基)对3种细胞均无抗肿瘤活性;化合物6c(喹啉环4位苯氨基上连接3个氟原子)对BEL-7402和A549细胞无活性,对SGC-7901细胞有较好的抑制效果(IC50为15.04 μmol/L),活性与于吉非替尼相当(IC50为 14.08 μmol/L);化合物6d(喹啉环4位为吡啶基)对SGC-7901和A549细胞均无活性,而对BEL-7402细胞有中等抑制作用(IC50为 49.77 μmol/L).
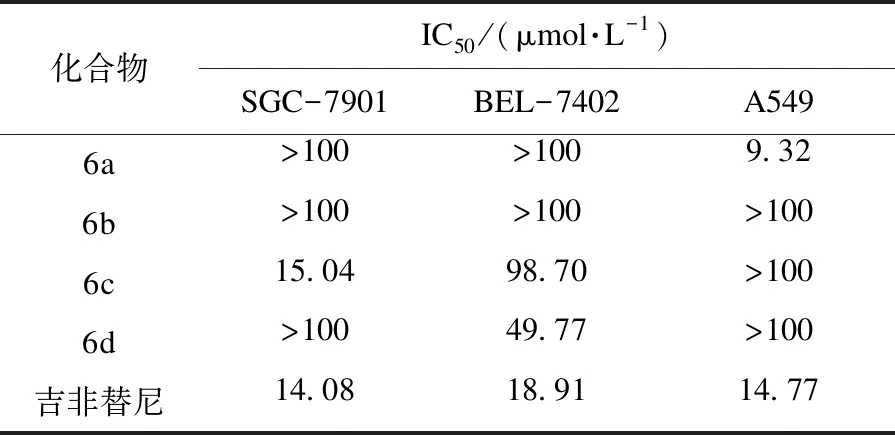
表1 目标化合物对人体癌细胞的体外抗增殖活性Table 1 In vitro anti-proliferative activity of target compounds against human cancer cell lines
3 结 论
实验设计并合成了4个未见报道的6-硝基-4-芳氨基喹啉化合物,测定了其对SGC-7901、BEL-7402和A549细胞的抗肿瘤活性.化合物6a对A549细胞的抑制效果最好,IC50为9.32 μmol/L,活性优于阳性对照药吉非替尼;化合物6c对SGC-7901细胞的抑制效果较好,与吉非替尼活性相当,IC50为15.04 μmol/L;6-硝基-4-芳氨基喹啉化合物进一步衍生化研究、抗肿瘤活性及构效关系研究正在进行中.
猜你喜欢
中国药理学与毒理学杂志(2022年7期)2022-10-17
云南化工(2022年2期)2022-03-18
动物营养学报(2022年1期)2022-02-20
热带农业工程(2017年2期)2017-08-29
吉林农业(2017年5期)2017-05-13
江苏农业科学(2015年1期)2015-04-17
今日农药(2014年7期)2014-09-15
中国医学创新(2013年2期)2013-03-28
