
不同种植地苜蓿根际土壤细菌群落结构多样性差异分析
2021-06-07 06:07翟亚萍王绍明
新疆农业科学 2021年5期
翟亚萍,王绍明,刘 鸯,杨 盼,张 霞,赵 祥,刘 丹
(石河子大学生命科学学院,新疆石河子 832000)
0 引 言
【研究意义】紫花苜蓿(MedicagosativaL.)种植产业增长空间较大[1]。研究土壤微生物多样性,群落结构、种类组成及其功能预测,有助于阐明生物多样性与生态系统功能之间的关系[2]。土壤细菌是土壤微生物中种类最多、数量最大、在土壤有机质分解、腐殖质形成、养分转化与吸收等过程中起到重要作用的类群,其群落结构组成及多样性变化是表征土壤环境质量的敏感指标[3-4]。【前人研究进展】李怡等[5]采用高通量测序技术分析了林地根际与非根际土壤细菌的物种组成、多样性和群落结构。赵祥等[6]研究表明,滴灌增加了植物根际土壤中细菌多样性和丰度。李文广等[7]合理复种饲料油菜并还田可提高后茬麦田土壤养分含量及酶活性,并有效改善细菌群落多样性。【本研究切入点】目前,新疆北疆苜蓿土壤微生物研究尚未见文献报道。研究新疆北部不同种植地苜蓿土壤细菌群落多样性及群落分布特征。【拟解决的关键问题】研究基于IonS5TMXL测序平台进行测序,Tax4Fun功能预测进行功能分类,对新疆天山北麓石河子地区、塔城地区苜蓿土壤细菌群落结构和多样性进行分析。分析细菌群落组成与土壤酶活性之间的关系,为研究新疆北部苜蓿土壤微生物的群落组成、物种多样性及生态功能提供科学依据。
1 材料与方法
1.1 材 料
研究区选取新疆天山北麓石河子地区、塔城地区种植苜蓿地,属于温带干旱大陆性气候区,冬季长而严寒,夏季短而炎热。石河子地区年平均气温7~8℃,北部地区气温低,南部高。日照2 300~2 700 h,年均降雨量180~270 mm,年蒸发量1 000~1 500 mm。塔城地区降水量稍多,年均降雨量 290 mm,年蒸发量1 600 mm,日照2 800~3 000 h,无霜期 130~190 d。
1.2 方 法
1.2.1 样品采集
2018年7月在新疆石河子地区,塔城地区选取6个不同种植苜蓿地作为样地,所取样品分为12种土壤类型。分别在6个样地内设置3个5 m×5 m的样方,每个样方内选取5~10株植株,根际土壤取样采用“抖根法”,植物根系从土壤中挖出,抖掉与根系结合松散的土壤,收集与根系紧密结合在 4 mm的土壤作为根际土壤,在采集根际土旁选取没有植物生长的位置采集与根际土相同深度的土壤作为非根际土壤样品。土壤于4℃保存并尽快带回实验室,1份保存于-80℃,供提取土壤 DNA 进行高通量测序;另1份风干保存,用于基础数据测定。12种土壤类型分别为紫泥泉苜蓿根际土壤(简化为ZR),紫泥泉苜蓿非根际土壤(ZN),147团苜蓿根际土壤(SR),147团苜蓿非根际土壤(SN),沙湾县苜蓿根际土壤(WR),沙湾县苜蓿非根际土壤(WN),额敏县苜蓿根际土壤(ER),额敏县苜蓿非根际土壤(EN),托里县苜蓿根际土壤(TR),托里县苜蓿非根际土壤(TN),裕民县苜蓿根际土壤(XR),裕民县苜蓿非根际土壤(XN)。表1

表1 采样地点及样品信息
1.2.2 土壤理化性质及酶活性
土壤理化指标的测定参考鲍士旦《土壤农化分析》中的测定方法。
1.3 数据处理
基于IonS5TMXL测序平台,利用单端测序(Single-End)的方法,构建小片段文库进行单端测序,测序得到的下机数据(Raw Data)进行OTUs聚类和物种分类分析。对OTUs进行丰度、Alpha多样性计算等分析,得到样品内物种丰富度和均匀度信息、不同样品或分组间的共有和特有OTUs信息等。将高通量测序获得的有效数据按照97%的一致性将序列聚类成为OTUs后,通过与数据库Silva132比对,进行物种注释,并对不同分类层级统计。进行组间差异的比较,揭示不同处理或环境下的群落结构的差异特征。将高通量测序获得的有效数据按照97%的一致性将序列聚类成为OTUs后,通过与数据库Silva132比对,进行物种注释,并对不同分类层级统计发现:共有7 343个OTUs,其中,能够注释到数据库的OTUs数目为7 336(99.90%)。
选取每个样本在门分类水平上最大丰度排名前10的物种,生成物种相对丰度柱形累加图。根据12个土样类型在属水平的物种注释及丰度信息,选取丰度排名前35的属,根据其在每个样本中的丰度信息,从物种和样本两个层面进行聚类,绘制成热图。Tax4Fun功能预测根据样品在数据库中的功能注释及丰度信息,基于KEGG PATHWAY数据库从功能差异层面进行聚类。
Tax4Fun功能预测是通过基于最小16S rRNA 序列相似度的最近邻居法实现的,提取KEGG数据库原核生物全基因组16S rRNA 基因序列并利用BLASTN 算法将其比对到SILVA SSU Ref NR 数据库(BLAST bitscore >1 500)建立相关矩阵, 通过UProC和PAUDA 2种方法注释的KEGG数据库原核生物全基因组功能信息对应到SILVA数据库中,实现SILVA数据库功能注释。测序样品以SILVA数据库序列为参考序列聚类出OTU,进而获取功能注释信息。
2 结果与分析
2.1 土壤营养成分
研究表明,石河子地区、塔城地区12种土壤类型均表现出根际土壤纤维素酶、脲酶和中性磷酸酶含量均高于非根际土壤,过氧化氢酶含量除托里县苜蓿根际(TR)与非根际(TN)外均表现出根际土壤高于非根际土壤,蔗糖酶含量除额敏县苜蓿根际(ER)与非根际(EN)外也均表现为根际土壤高于非根际土壤。其中除托里县苜蓿根际(TR)与非根际(TN)过氧化氢酶含量和额敏县苜蓿根际(ER)与非根际(EN)蔗糖酶含量外各组根际土壤与非根际土壤5种酶活性均呈现显著性差异。表2
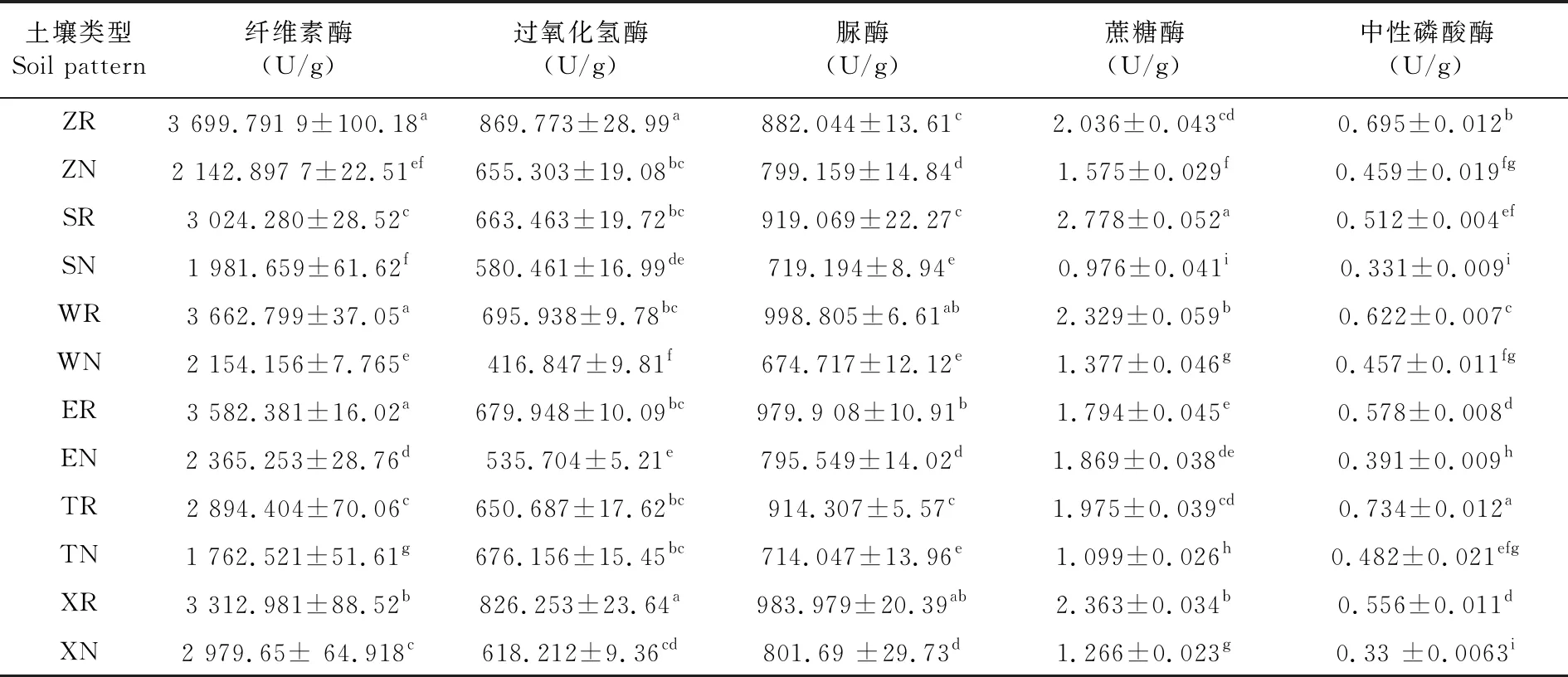
表2 不同土样类型下土壤酶活性
2.2 样品间 Alpha 多样性
研究表明,对36个土壤样品进行高通量测序经方法后共获得2 581 720条高质量序列,平均长度为412 nt。各样品文库的覆盖率(Coverage)均大于97.8%,各样地的微生物物种信息得到了充分的体现,测序结果能够真实的反映出土壤细菌群落的情况。Shannon指数、Simpson指数表现为过氧化氢酶含量较高样地(TR、SR、ER)>过氧化氢酶含量较低样地(WR、XR、ZR),Chao1指数和Ace指数表现为中性磷酸酶含量较高样地(SR、XR、ER)>中性磷酸酶含量较低样地(WR、ZR、TR)。其中Simpson指数除额敏县苜蓿根际(ER)与非根际(EN)外各组根际与非根际之间没有显著性差异,Chao1指数除紫泥泉苜蓿根际(ZR)与非根际(ZN)外各组根际与非根际之间没有显著性差异。表3

表3 细菌群落多样性指数
2.3 土壤细菌多样性指数与土壤酶活性之间的关系
研究表明,中性磷酸酶与Chao1指数、Ace指数呈显著正相关关系;过氧化氢酶与Shannon指数、Simpson指数、Chao1指数和Ace指数呈负相关;Simpson指数与纤维素酶、脲酶和中性磷酸酶呈负相关关系;蔗糖酶与Shannon指数、Simpson指数、Chao1指数和Ace指数呈正相关关系。表4

表4 土壤细菌多样性指数与土壤酶活性之间的相关性
2.4 细菌群落结构多样性
研究表明,共有 7,343个OTUs,其中,能够注释到数据库的OTUs数目为7,336(99.90%),注释到界水平的比例为99.90%,门水平的比例为91.47%,纲水平的比例为79.12%,目水平的比例为66.27%,科水平的比例为52.62%,属水平的比例为33.45%,种水平的比例为7.42%。
在门水平上,所测12个土样类型的序列共分属于51个门,共同拥有的有22个门,排名前10的分别为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、芽单胞菌门(Gemmatimonadetes)、绿弯菌门(Chloroflexi)、疣微菌门(Verrucomicrobia)、拟杆菌门(Bacteroidetes)、广古菌门 (Euryarchaeota) 、浮霉菌门 (Planctomycetes)和 厚壁菌门 (Firmicutes) 。门分类水平上,12个土样类型细菌群落结构大致相同,优势种群相似但丰度略不相同。图1

图1 不同土样类型下细菌在门分类水平上的群落结构
研究表明,在属分类水平上,测得12个土样类型的序列共分属于600个属;其中12个土样类型共同拥有的有190个属,在群落中占比例较高的有10个属,分别寡养单胞菌属 (Stenotrophomonas)、鞘氨醇单胞菌属(Sphingomonas) 、芽球菌属( Blastococcus) 、节杆菌属 (Arthrobacter) 、红色杆菌属(Rubrobacter)、unidentified_Acidobacteria、unidentified_Actinomarinales、慢生根瘤菌(Bradyrhizobium)、Gaiella、土壤红杆菌属(Solirubrobacter)。图2
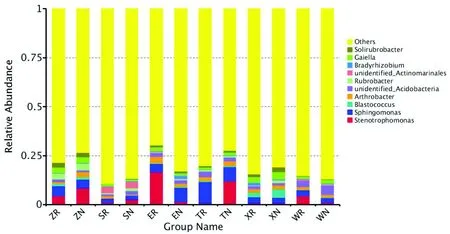
图2 不同土样类型下细菌在属分类水平上的群落结构
聚类主要集中在变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、芽单胞菌门(Gemmatimonadetes)、疣微菌门(Verrucomicrobia)和拟杆菌门(Bacteroidetes)6个门类中。各样地根际与非根际相比:紫泥泉苜蓿根际(ZR)提高了Gaiella、土壤红杆菌属(Solirubrobacter)、红色杆菌属(Rubrobacter)、Pelagibius的丰度;147团苜蓿根际(SR)的优势菌属为藤黄单胞菌属(Luteimonassp)、Pelagibius;沙湾县苜蓿根际(WR)提高了Polycyclovorans的丰度;额敏县苜蓿根际(ER)的优势菌属为寡养单胞菌属 (Stenotrophomonas) 、节杆菌属 (Arthrobacter) ;托里县苜蓿根际(TR)的优势菌属为鞘氨醇单胞菌属(Sphingomonas)、Stenotrophobacter、新鞘氨醇杆菌属(Novosphingobium);裕民县苜蓿根际(XR)提高了红游动菌属 (Rhodoplanes)、慢生根瘤菌(Bradyrhizobium)丰度。图3
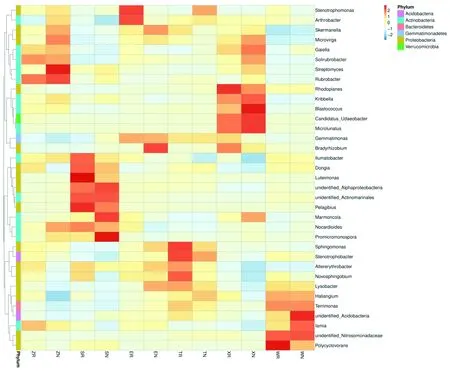
图3 不同土样类型下细菌在属分类水平上的热聚类
2.5 土壤细菌群落功能预测
研究表明,紫泥泉苜蓿根际(ZR)中遗传信息处理(Genetic_Information_Processing)、有机体系统(Organismal_Systems)功能丰度较高;147团苜蓿根际(SR)丰度较高的功能预测为新陈代谢(Metabolism)、未分类(Unclassified)、人类疾病(Human_Diseases);沙湾县苜蓿根际(WR)环境信息处理(Environmental_Information_Processing)、细胞过程(Cellular_Processes)、未分类(Unclassified)、人类疾病(Human_Diseases);额敏县苜蓿根际(ER)中细胞过程(Cellular_Processes)、人类疾病(Human_Diseases)功能丰度较高;托里县苜蓿根际(TR)环境信息处理(Environmental_Information_Processing)、细胞过程(Cellular_Processes)、人类疾病(Human_Diseases)的丰度提高;裕民县苜蓿根际(XR)比非根际(XN)新陈代谢(Metabolism)、未分类(Unclassified)的功能丰度提升。图4
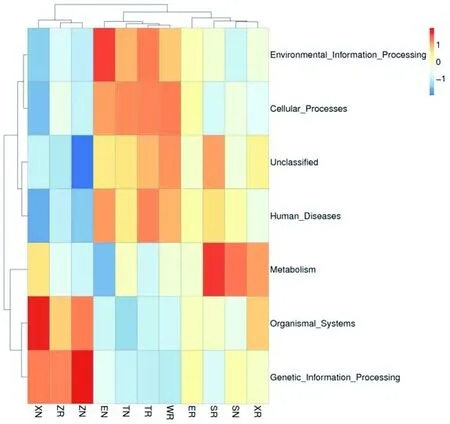
图4 不同土样类型细菌功能预测热聚类
3 讨 论
研究表明,种植苜蓿能够明显提高土壤酶活性,而土壤酶活性是影响土壤中微生物群落组成的因素之一[8]。土壤脲酶能够提高土壤氮素的生物有效性;土壤蔗糖酶参与蔗糖降解;土壤中性磷酸酶催化有机磷水解, 供植物吸收;土壤过氧化氢酶可以分解土壤中对植物有害的自由基—过氧化氢;纤维素酶可以分解纤维素和木质素。研究细菌群落丰富度指数(Chao1指数和Ace指数)与土壤中性磷酸酶呈显著正相关,说明细菌能够提高土壤中性磷酸酶含量并且促进根际土壤磷养分的累积,而土壤中性磷酸酶的提升能够增加细菌群落多样性。石河子地区、塔城地区苜蓿根际土壤有着丰富的微生物资源,通过对样品间 Alpha 多样性分析可知,石河子地区3个样地相较于塔城地区3个样地细菌群落多样性较高,但是丰富度较低。石河子地区盐碱化程度严重及土地熟化程度不及塔城地区,细菌种类总数较低,不同环境条件下石河子地区、塔城地区苜蓿根际土壤的细菌优势种群不尽相同。
在不同的12个土壤样本中,丰度最高的为变形菌门(Proteobacteria)和放线菌门(Actinobacteria),这与前人研究结果一致[3]。其中放线菌门、变形菌门中的部分微生物参与土壤有机质矿化, 具有分解纤维素、半纤维素、蛋白质和木质素等功能[9],这证实了研究中土壤纤维素酶、土壤蔗糖酶与细菌群落多样性指数呈正相关。中性磷酸酶含量较高的样地(SR、XR、ER)中丰度较高的为藤黄单胞菌属(Luteimonassp) 为近年来的新发现属,且尚未有其可产抗菌活性物质的报道[10];Pelagibius为嗜盐细菌,能够适应盐化生境[11];红游动菌属 (Rhodoplanes) 可能是引起西洋参根腐病发生的关键菌群[12];慢生根瘤菌(Bradyrhizobium)能够利用糖类及有机酸,但更倾向于利用戊糖,前人研究发现该属细菌对抗生素的抗药性比固氮根瘤菌强[13];寡养单胞菌属 (Stenotrophomonas)可以降解有机农药 、进行废水处理和污染水体的修复、部分寡养单胞菌对农药具有生物转化活性[14];节杆菌属 (Arthrobacter) 具有很强的环境适应性和抗逆性,研究发现该属菌株具有高效降解环境中的有机污染物和吸附重金属的能力,被用于污染水体和土壤等的生物修复研究与应用[15]。中性磷酸酶含量较低的样地中丰度较高的为(WR、ZR、TR)红色杆菌属(Rubrobacter)是对于适应沙漠干旱、强辐射等极端环境具有重要作用[16];新鞘氨醇杆菌属(Novosphingobium) 普遍具有降解芳烃 (芳香族) 化合物的特性是良好的芳烃污染环境的生物修复菌[17]。
在新疆北部不同样地的苜蓿根际细菌群落一级功能层中, 中性磷酸酶含量较高的样地(SR、XR、ER)中丰度较高的有机体系统、新陈代谢、遗传信息处理这3类功能基因使土壤细菌代谢旺盛,从土壤中吸收营养物质,通过摄取氨基酸、能量、碳水化合物等来保证细菌的存活从而提高了细菌群落结构的多样性[18];中性磷酸酶含量较低的样地(WR、TR)中环境信息处理、细胞过程功能基因丰度较高,通过这些功能基因可以从土壤中摄取更多的核苷酸、氨基酸,提高了细菌多样性[19]。
4 结 论
细菌多样性主要受土壤中性磷酸酶影响,根际与非根际及不同地区间,所测12个土样类型的序列共分属于51个门,共同拥有的有22个门,共分属于600个属;其中12个土样类型共同拥有的有190个属,细菌群落组成相似,但物种丰富度存在一定差异。土壤细菌群落功能预测中一级功能层中遗传信息处理(Genetic_Information_Processing)、有机体系统(Organismal_Systems)、新陈代谢(Metabolism)、未分类(Unclassified)、人类疾病(Human_Diseases)、环境信息处理(Environmental_Information_Processing)、细胞过程(Cellular_Processes)丰度较高。
猜你喜欢
中国土壤与肥料(2022年4期)2022-06-14
济南大学学报(自然科学版)(2022年3期)2022-05-18
林业科学(2022年2期)2022-05-11
草地学报(2022年4期)2022-04-25
河海大学学报(自然科学版)(2022年1期)2022-02-11
作物学报(2022年3期)2022-01-22
四川农业科技(2021年7期)2021-12-01
河南农业·综合版(2020年9期)2020-09-27
分析化学(2018年7期)2018-09-17
天津农业科学(2014年12期)2014-12-11
