
Rnd3表达改变对糖尿病大鼠内皮祖细胞生物学特性的影响*
2021-06-02 11:55张书娅邵钟铭伍彩霞刘晓晓郭峻莉
中国病理生理杂志 2021年5期
张书娅,邵钟铭,伍彩霞,刘晓晓,邹 园,揭 伟,△,郭峻莉△
(1海南医学院第一附属医院海南省热带心血管病研究重点实验室,海南医学院急救与创伤研究教育部重点实验室,海南海口571199;2广东医科大学基础医学院病理学系,广东湛江524023)
当前全球糖尿病患病率增长迅速且居高不下。据统计,2019年糖尿病患病数仅在20~79岁年龄段全世界就有4.63亿;预计到2045年,这个数字将会增加超过7亿[1]。血糖持续控制不良的情况下,糖尿病血管并发症的患病率、致残率和致死率也相应增加。血管功能障碍在糖尿病血管并发症易感性方面起着重要作用,也是独立预测因子[2]。内皮祖细胞(endothelial progenitor cells,EPCs)是内皮细胞的前体细胞,并且是多能干细胞,在再生医学研究中具有重要的地位[3]。糖尿病状态下EPCs数量和功能受损是糖尿病血管并发症发生的重要机制。在血管受到缺血或缺氧刺激时,骨髓来源的EPCs可从骨髓动员到外周血并趋化迁移至缺血损伤部位分化为成熟的血管内皮细胞,形成新生血管,直接发挥其血管新生作用,也可通过分泌促血管新生的细胞因子以“旁分泌”的形式间接发挥其促血管新生的作用[4-5]。因而,EPCs的数量及增殖、迁移和分化等功能与血管疾病进程和死亡率呈负相关[6-7]。
Rho家族GTP酶3(Rho family GTPase 3,Rnd3)是小GTP酶Rho家族的Rnd亚家族成员[8-9],可以竞争性结合RhoA的靶分子ROCK1而拮抗RhoA的生物学功能,在细胞的骨架和极性维持、周期、凋亡、迁移及黏附过程中起到了关键作用[10-13]。早期有研究显示[14],与消瘦的非糖尿病人群相比,伴有肥胖的Ⅱ型糖尿病患者的骨骼肌组织中Rnd3表达增加,ROCK1活性减少,初步提出了糖尿病状态与Rnd3表达之间的关系。本课题组前期结果显示,Rnd3表达不足抑制了心肌梗死后血管新生,因而是体内调控血管新生的重要因子[15]。关于Rnd3与EPCs功能的关系,特别是在糖尿病状态下的影响,目前尚未见报道。本研究旨在应用Rnd3基因敲除的大鼠,首次探讨糖尿病状态下Rnd3改变对骨髓源性EPCs一般生物学特性的影响,为后续相关机制和在体研究提供实验依据和理论基础。
材料和方法
1 实验动物
6周龄雌性和雄性Rnd3基因敲除的杂合子(Rnd3+/-)Sprague-Dawley(SD)大鼠均由江苏赛业生物科技有限公司提供,许可证号为SYXK(粤)2015-0147。Rnd3+/-大鼠饲养并繁殖于广东医科大学医学实验动物中心,子代鼠经基因型分析,分别得到Rnd3+/-和野生型(wild-type,WT)大鼠。
2 材料和主要试剂
RPMI-1640培养基和链脲佐菌素(Sigma);EGM-2专用培养基(LONZA);FITC-UEA-1和胎牛血清(fetal bovine serum,FBS)购自Gibco;大鼠淋巴细胞分离液(天津灏洋生物公司);DiI标记的乙酰化低密度脂蛋白(DiI-acLDL)购自广州奕源生物科技有限公司;抗β-actin抗体(Santa Cruz);抗caspase-3抗体(CST);抗Bcl-2抗体(Proteintech);增强型CCK-8试剂盒(碧云天);Matrigel和Transwell小室(Corning);血管内皮生长因子(vascular endothlial growth factor,VEGF)ELISA试剂盒(R&D)。
3 实验方法
3.1 糖尿病SD大鼠模型建立与分组委托江苏赛业生物公司基于CRISPR/Cas9技术构建Rnd3基因敲除SD大鼠,经PCR和测序鉴定合格的Rnd3+/-大鼠用于后代繁育,得到Rnd3+/-和WT大鼠。分别使用成年Rnd3+/-和WT大鼠来构建糖尿病动物模型,参考既往报道进行[16]。简言之,使用高脂饲料适应性喂养大鼠1周。将STZ与柠檬酸钠缓冲液混匀,配制终浓度为10 g/L。实验组予STZ溶液以60 mg/kg一次性腹腔注射,对照组注射等比例柠檬酸钠缓冲液。期间饲以高脂饲料。STZ注射3周后,以连续2次空腹血糖值≥16.7 mmol/L为建模成功。实验分为4组:WT对照(WT-Ctrl)组、WT糖尿病(WT-DM)组、Rnd3基因敲除杂合子对照(Rnd3+/--Ctrl)组和Rnd3基因敲除杂合子糖尿病(Rnd3+/--DM)。每组20只大鼠。
3.2 大鼠骨髓源性EPCs的分离、培养及鉴定采用既往报道方法进行[17]。简言之,大鼠经阿佛汀完全麻醉后,将双下肢迅速剥离,注意保持完整性。使用RPMI-1640培养基反复冲洗骨髓腔,经70μm滤网过滤。采用大鼠淋巴细胞分离液分离出骨髓单个核细胞,RPMI-1640培养基清洗1次。将得到的细胞加入EGM-2内皮细胞专用完全培养基,轻轻重悬底层细胞。培养皿放入37℃、5%CO2培养箱静置培养。48 h后吸取上清,二次接种于新的培养皿中继续培养。以后每48~72 h用EGM-2完全培养基换液。收集第3代贴壁细胞,加入10 mg/L的DiI-acLDL,避光于37℃孵育4 h;PBS漂洗2次,4%多聚甲醛固定20 min;加入10 mg/L的FITC-UEA-1,避光孵育1 h,PBS漂洗3次,加入抗荧光淬灭封片液封片。使用激光扫描共聚焦显微镜鉴定。DiI-acLDL和FITC-UEA-1表达双阳性可以鉴定为EPCs[18]。本实验所用的EPCs未做纯度检测。
3.3 CCK-8法检测细胞活力取WT-Ctrl、WT-DM、Rnd3+/--Ctrl和Rnd3+/--DM组生长状态良好的第2代EPCs以每孔3×103接种于96孔板,每组6个复孔。每孔加入10μL CCK-8试剂,37℃、5%CO2培养箱中继续孵育4 h,酶标仪上读取450 nm的吸光度(A)值。共设置0 d、1 d、2 d、3 d、5 d和7 d共6个时点。
3.4 Western blot检测各组EPCs的细胞凋亡相关分子的表达分别提取各组第2代EPCs总蛋白,BCA法测定蛋白浓度,每孔按35μg蛋白上样,经12%的SDS-PAGE分离。蛋白转移至NC膜,各膜经含5%BSA的TBST室温摇床上封闭2 h,TBST洗涤2次后与相应Ⅰ抗过夜4℃孵育。抗体使用情况如下:Bcl-2,1∶500;caspase-3,1∶1 000;β-actin,1∶2 000。TBST洗涤15 min×3遍,各膜再与HRP-标记IgG室温下结合1 h,洗涤后经ECL发光液显影,天能凝胶成像分析系统扫描和分析条带。
3.5 Transwell小室细胞迁移实验第2代细胞以无血清同步化2 h,各组细胞以每孔2×104的数量(总体积200μL,含0.5%FBS)接种于Transwell小室的上室,下室每孔加入500μL含10%FBS的EGM-2完全培养液,细胞常规培养24 h后取出小室,PBS清洗小室2遍,用棉签轻轻擦去微孔膜上层的细胞。4%多聚甲醛固定20 min后,结晶紫染色30 min,三蒸水漂洗3次,晾干,倒置显微镜观察,拍照计数。每组设置3个复孔。
3.6 ELISA检测EPCs上清液中VEGF含量分别将同样数量的对数生长期的第2代EPCs接种于6孔板,收集24 h贴壁后各孔内细胞上清标记为0 d组,细胞继续培养3 d后再次收集未换液的细胞上清,标记为3 d组。根据ELISA试剂盒配制所有试剂与样品进行VEGF浓度检测。每组3个复孔。
3.7 EPCs成管实验48孔板每孔加入100μL稀释后的Matrigel,放入37℃培养箱孵育30 min。第2代EPCs用无血清EBM-2培养基饥饿处理2 h,以每孔2×104的密度接种于Matrigel上,37℃、CO2培养箱培养18 h后,置于光镜下观察,拍照,使用ImageJ软件处理图片。
4 统计学处理
采用GraphPad Prism 8.0软件进行统计学分析,数据以均数±标准差(mean±SD)表示。采用单因素方差分析进行Tukey两两对比分析,以P<0.05为差异有统计学意义。
结 果
1 成功分离大鼠骨髓源性EPCs
分离的骨髓单个核细胞经EGM-2培养基选择性培养7 d后,可观察到细胞呈簇状,周围呈梭形为主并放射状生长,集落中央的细胞呈圆形,细胞较小。继续培养至21 d左右,细胞形态相互融合呈典型的“铺路石”状(图1A)。在荧光显微镜下观察,吞噬DiI-acLDL的细胞呈现红色荧光标记,特异结合FITC-UEA-1的细胞呈绿色荧光,同时摄取DiI-acLDL又结合FITC-UEA-1呈橘黄色荧光标记的双阳性即为EPCs(图1B)。
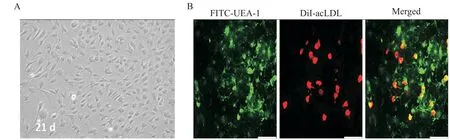
Figure 1.The morphological and functional identification of EPCs.A:morphological change of EPCs at 21 d;B:functional identification of EPCs under fluorescence microscope.The scale bar=150μm.图1 EPCs的形态与功能鉴定
2 糖尿病状态增强了Rnd3基因敲除对EPCs的活力的抑制效应
CCK-8实验结果显示,相对于WT-Ctrl组,WTDM组在3 d时细胞活力显著下降(P<0.05),Rnd3+/--Ctrl组在5 d时显示出显著下调趋势(P<0.05)。与WT-Ctrl和Rnd3+/--Ctrl组对比,Rnd3+/--DM在各时点均显示了显著的下降趋势(P<0.05),见图2。
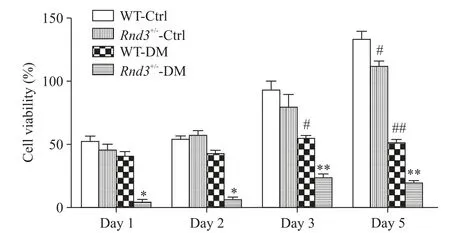
Figure 2.Insufficient expression of Rnd3 promoted the inhibitory effect of diabetes on the viability of EPCs.Mean±SD.n=3-6.*P<0.05,**P<0.01 vs WT-DMgroup;#P<0.05,##P<0.01 vs WT-Ctrl group.图2 Rnd3表达不足促进了糖尿病对EPCs活性的抑制效应
3 糖尿病状态下Rnd3基因敲除改变EPCs中细胞凋亡相关蛋白的表达
使用Western blot检测各组EPCs中凋亡相关蛋白Bcl-2和caspase-3的表达,结果如图3所示。凋亡抑制蛋白Bcl-2在Rnd3+/--Ctrl组与Rnd3+/--DM组相较于WT-Ctrl组EPCs中的表达都表现为下降,而促进凋亡蛋白总caspase-3则表达上调(P<0.05,P<0.01)。本研究中未检测活化的caspase-3蛋白。
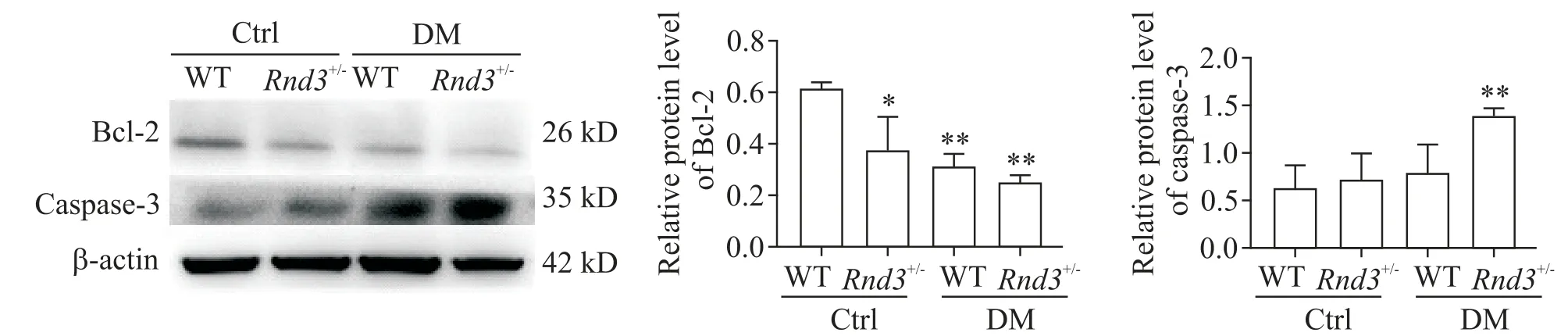
Figure 3.Insufficient expression of Rnd3 changed the expression of apoptosis-related proteins in EPCs from diabetic rats.Mean±SD.n=3.*P<0.05,**P<0.01 vs WT-Ctrl group;#P<0.05 vs WT-DM group.图3 Rnd3表达不足改变了糖尿病对EPCs凋亡相关蛋白的表达
4 糖尿病状态下Rnd3敲除抑制EPCs的细胞迁移能力
Transwell小室迁移实验结果显示(图4),相对于WT-Ctrl组,WT-DM和Rnd3+/--Ctrl组穿过PVDF膜的细胞数明显下降(P<0.05);与Rnd3+/--Ctrl组对比,Rnd3+/--DM组迁移能力进一步下降(P<0.05)。
5 糖尿病状态下Rnd3敲除抑制EPCs分泌VEGF
应用ELISA检测各组EPC释放至培养液上清中VEGF浓度的结果显示,与WT-Ctrl组相比较,WTDM组和Rnd3+/--Ctrl组EPCs释放VEGF水平均明显减弱,提示糖尿病和Rnd3表达减弱均显著抑制了EPCs释放VEGF;与Rnd3+/--Ctrl组相比,Rnd3+/--DM组3 d时VEGF浓度与Rnd3+/--Ctrl组相似,均处于与0 d时相似的基线水平,已无进一步下调的空间(图5)。
6 糖尿病状态下Rnd3敲除减弱了EPCs的血管样结构形成能力
成管实验结果显示,糖尿病显著抑制了EPCs的血管样结构形成能力,而Rnd3敲除则进一步加剧了糖尿病对EPCs血管样结构形成能力的抑制效应(图6)。
讨 论
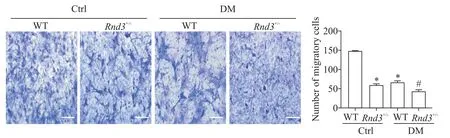
Figure 4.Defects in Rnd3 expression amplified the inhibitory effect of diabetes on the migration of EPCs.The scale bar=200μm.Mean±SD.n=3.*P<0.05 vs WT-Ctrl group;#P<0.05 vs Rnd3+/--Ctrl group.图4 Rnd3敲除放大了糖尿病对EPCs迁移的抑制效应
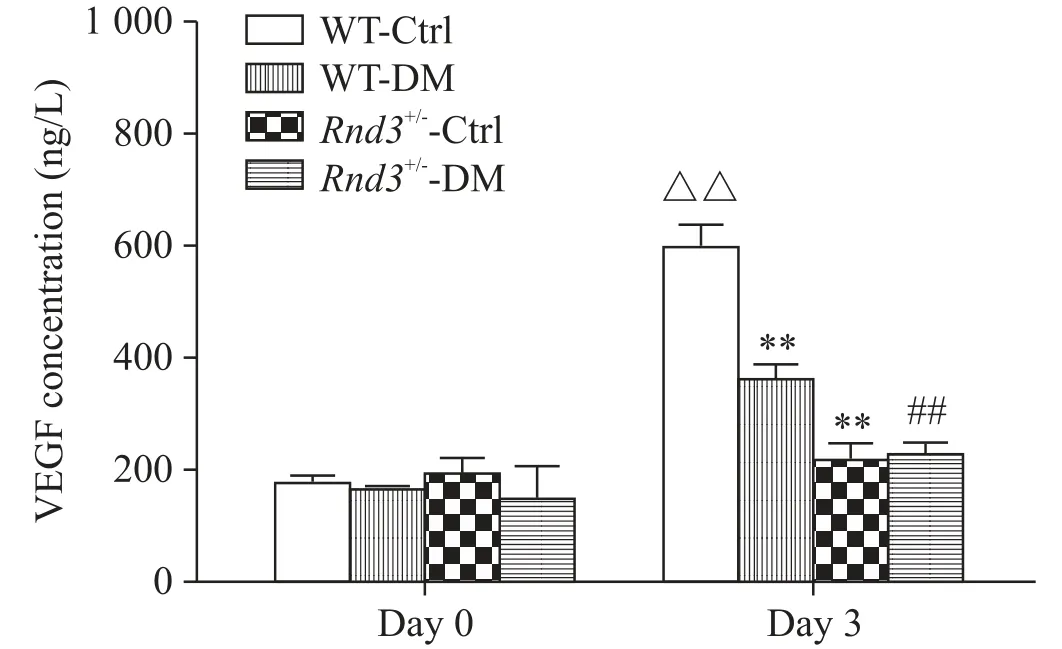
Figure 5.Insufficient expression of Rnd3 deteriorated the inhibitory effect of diabetes on the secretion of VEGF in EPCs(2×105 cells).Mean±SD.n=6.**P<0.01 vs WT-Ctrl group;##P<0.01 vs WT-DMgroup;△△P<0.01 vs day 0.图5 Rnd3敲除恶化了糖尿病对EPCs中VEGF分泌的抑制作用
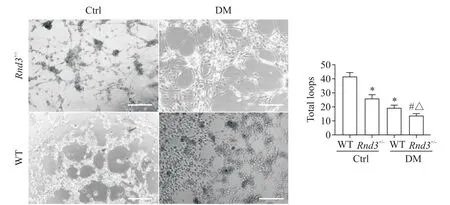
Figure 6.Insufficient expression of Rnd3 deteriorated the inhibitory effect of diabetes on the ability of EPCs to form tube-like structures.The scale bar=200μm.Mean±SD.n=3.*P<0.05 vs WT-Ctrl group;#P<0.05 vs Rnd3+/--Ctrl group;△P<0.05 vs WT-DMgroup.图6 Rnd3敲除恶化了糖尿病对EPCs血管样结构形成能力
血管内皮细胞功能紊乱是糖尿病血管并发症的根本原因。糖尿病伤口局部血管新生及伤口愈合与EPCs的数量及功能密切相关[19-20]。已经证实,无论是I型糖尿病还是II型糖尿病患者,其外周血EPCs的数量和血管生成功能均显著下降[21-22],但相关的机制仍不明确。因而,明确糖尿病患者EPCs数量减少及功能受损的机制,从而寻找合适靶点来改善患者自体EPCs增殖和迁移等功能,并提高其分泌促血管新生因子的能力,可能为将来EPCs移植治疗糖尿病缺血性心血管病提供重要的理论基础。
预实验发现,糖尿病状态下野生型EPCs要比正常状态下EPCs中Rnd3蛋白的表达减少(未发表资料),同时鉴于前期观察到Rnd3表达不足后显著抑制了心肌梗死后的血管生成能力[15],我们设想Rnd3不足也可能影响了EPCs的功能。为此,本研究基于CRISPR/Cas9技术编辑了大鼠Rnd3基因,获得Rnd3基因敲除后的杂合子和WT鼠,复制了糖尿病模型,分离了骨髓源性EPCs,首次探索Rnd3不足对EPCs一般生物学特性的影响。实验结果显示,糖尿病状态明显减弱了野生型SD大鼠骨髓源性EPCs的活力、迁移和旁分泌VEGF能力,促进细胞凋亡的蛋白表达增加,功能上表现为成血管能力的削弱。这些结果与既往的他人报道的有关糖尿病损伤EPCs的基本生物学功能的结果高度一致[23-25]。本实验发现Rnd3表达与既往Chun等[14]的报道的在糖尿病患者骨骼肌中Rnd3表达上调的结果相反,其原因可能为Chun等[14]的报道纳入对象为消瘦非糖尿病、肥胖非糖尿病和肥胖伴糖尿病三类人群,肥胖伴糖尿病者与消瘦非糖尿病人群对比,组织中Rnd3表达是升高的;而本研究构建的糖尿病大鼠是消瘦状态。两者实验对象的差异可能是Rnd3表达趋势相反的根本原因。本研究中我们首次发现,Rnd3表达不足明显的增加了糖尿病对EPCs上述活力、迁移、旁分泌和血管生成等一般生物学特性的负性影响作用,但相关机制尚不明确。
Rnd3是缺乏GTP水解活性的非典型Rho GTP酶,该分子具有多种细胞生物学功能,在细胞增殖、周期、凋亡、迁移、极性化及分化中发挥重要作用[26-28]。Rnd3不足可影响HIF-1α的稳定性而直接抑制HIF-1α下游靶基因VEGF的表达[15]。Rnd3缺陷也可通过影响p65和p50的稳定性而促进NF-κB信号的活化[29]。早期的报道显示糖尿病心肌组织中HIF-1α表达下调而NF-κB信号是活化的[30-31],由此,我们猜测本研究发现的Rnd3不足导致EPCs相应生物学功能的缺陷很可能也与HIF-1α和NF-κB信号的异常活化有关。生物信息学分析表明,大鼠Rnd3基因启动子-2010~120 bp上具有3个低氧反应元件,我们猜测Rnd3与HIF-1α之间极有可能形成信号调控反馈环路。因此很可能糖尿病时此调控反馈环路异常调节是Rnd3参与调控EPCs生物学特性的重要机制之一,但这一猜测还需进一步研究来证实。
总之,本研究基于Rnd3基因编辑大鼠成功复制了II型糖尿病模型,首次发现Rnd3表达不足促进了糖尿病对骨髓源性EPCs的活力、迁移、血管生成能力的负性影响,因而维持Rnd3表达在防止糖尿病血管并发症上具有潜在的靶向干预价值。本研究未做EPCs纯度分析,不能排除其中含有其他细胞的可能和由此对结果可靠性和结论可信度的影响。尽管为了保证EPC的质量,我们在实际操作中是尽量用前2代细胞,以上结论仍需更多实验加以验证。
猜你喜欢
中国临床医学影像杂志(2021年10期)2021-11-22
国际放射医学核医学杂志(2020年3期)2020-07-27
文苑(2018年18期)2018-11-08
西江月(2018年5期)2018-06-08
海峡姐妹(2017年5期)2017-06-05
现代检验医学杂志(2016年2期)2016-11-14
中央民族大学学报(自然科学版)(2016年3期)2016-06-27
广西林业科学(2016年2期)2016-03-20
中国医药生物技术(2015年4期)2015-12-26
中国水利(2015年14期)2015-02-28

- 中国病理生理杂志的其它文章
- ERK信号通路介导的EP300过表达在苯肾上腺素诱导小鼠心肌细胞肥大中的作用*
- 益肾通络方对大鼠勃起功能障碍及NO-cGMP通路的影响*
- 静脉注射SCAD重组腺病毒减轻自发性高血压大鼠心肌肥厚和纤维化*
- Thioredoxin-interacting protein contributesto cardiac fibrosisby elevating oxidative stress in cardiac fibroblasts*
- 延髓头端腹内侧部NADPH氧化酶2激活导致活性氧簇释放在皮肤/肌肉切开和牵拉引起的慢性术后疼痛中的作用*
- 干扰海马spastin表达通过抑制突触传递介导小鼠认知功能障碍*