
基于晶体学原理的高效光催化材料的设计与制备
2021-05-20 10:08王泽岩刘媛媛郑昭科程合锋黄柏标
人工晶体学报 2021年4期
王泽岩,王 朋,刘媛媛,郑昭科,程合锋,黄柏标
(山东大学,晶体材料国家重点实验室,济南 250100)
0 引 言
自从1972年日本科学家Fujishima等[1]用TiO2单晶实现光催化分解水产生氢气和氧气以来,光催化技术吸引了来自世界各国科学家们的广泛关注。光催化技术是基于半导体材料的光电效应,可以将光能转化为化学能的新技术。当半导体材料吸收光子能量之后,产生电子和空穴,随后电子和空穴可分别发生还原和氧化反应,从而将光能转化为化学能。利用该技术,可实现光催化分解水产氢、二氧化碳还原制备有机物及将环境中的有机污染物降解为二氧化碳和水,而整个过程中不会产生任何二次污染。因此,光催化技术被认为是解决未来能源短缺和环境污染问题最具潜力的解决方案之一[2-3]。随着材料科学和纳米技术的不断发展,在过去几十年的时间里,光催化技术获得了快速的发展。目前人们已经研究出了几百种新型的光催化材料,材料的光催化效率也在不断提高。然而,目前光催化材料的能量转换效率距离实际应用的要求依然有一定距离。因此,进一步提高光催化材料的能量转换效率,探索新型高效的光催化材料依然是光催化领域的研究热点。
光催化反应的过程总体来讲可分为三个步骤:(1)半导体光催化材料吸收超过其能带宽度的光子能量产生电子和空穴;(2)光生电子和空穴产生后在光催化材料内部逐渐向材料表面迁移;(3)扩散到材料表面的电子和空穴分别同反应物发生还原和氧化反应。因此,光催化材料的能量转换效率总体来讲是由光催化材料的光吸收效率、光生载流子的内部分离效率和界面注入效率决定。针对光催化反应的三个过程,人们发展出各种各样的策略以进一步提高光催化材料的能量转换效率。例如:通过在半导体能带中引入杂质能级以拓展光催化材料的光吸收范围、将具有合适能带结构的半导体材料复合构建异质结以促进光催化材料内部光生电子和空穴的有效分离、在半导体光催化材料表面负载助催化剂以促进光催化材料表面电荷的分离和促进表面氧化还原反应进行等。然而,这些传统方式在解决某一方面问题的同时,将会引起其他方面的问题。例如:通过掺杂可在一定程度拓展光催化材料的光吸收范围,从而提高吸收的光子能量。然而杂质离子的引入可成为光生电子和空穴的复合中心,从而降低光生载流子的分离效率。通过材料复合构建 Ⅱ 型异质结,可在复合材料界面构建界面电场,从而为光生载流子分离提供驱动力,提高载流子分离效率,然而,界面的存在同样会产生大量缺陷,从而使界面成为光生载流子的复合中心。因此,发展提高光催化材料效率的新策略,探索具有更高光催化活性的新型光催化材料,对推动光催化技术发展及其实际应用具有重要的意义[4-6]。
材料的物理性能从本质上讲同其本身的晶体结构密切相关,不同晶体材料由于其内部原子排布的方式不同,其电子结构、物理性质也随之不同。因此,从晶体学基本原理出发,基于材料结构与性能的关系,通过对材料晶体结构、电子结构、微结构参数进行设计和调控,有望从根本上了解制约光催化材料活性的原因,从而探索具有较高光催化活性的新型光催化材料,并获得系列不同于传统提升半导体材料光催化活性的新策略。结合近期本课题组在新型高效光催化材料的设计与制备方面的研究结果,本文对其进行了简单的总结,期望对促进光催化技术的进一步发展,探索新型高效光催化材料,推动光催化技术应用提供一些启发。
1 表面等离子体光催化材料
表面等离子体光催化剂是一种具有贵金属表面等离子体共振效应的复合半导体光催化剂,体系中通常包含金属纳米颗粒和半导体材料,同时具有贵金属纳米颗粒(NPs)、半导体,以及复合光催化剂的优点,被认为是有望解决人类社会面临的能源、环境和医疗等问题的优异材料[1,2,7-10]。人们可以通过调控金属纳米颗粒的大小、形貌以及所处环境来调控其对光的吸收,因此表面等离子体光催化剂能够有效拓宽体系的光吸收;贵金属由于具有较低的费米能级,利于分离体系中所产生的光生电子和空穴;与此同时,贵金属纳米颗粒具有较高的催化活性,可以作为催化反应的活性位点,利于催化反应的进行。综上所述,表面等离子体光催化剂能够有效拓宽体系的光吸收,利于分离体系中光生载流子,同时能够促进催化反应的进行。科研人员目前已经开展了表面等离子体光催化剂方面的广泛研究,其中最为常见的表面等离子体光催化剂是贵金属纳米颗粒沉积在半导体或绝缘体颗粒的表面[11-12]。在金属-半导体复合光催化剂中,贵金属纳米颗粒由于其表面等离子体共振效应而成为捕获可见光的主要成分,而金属-半导体界面则有效地分离光激发产生的电子和空穴[10-12]。因此,表面等离子体光催化剂可以实现直接利用太阳能分解水产氢、降解污染物和转化CO2等[7-9,11-18]。不仅如此,表面等离子体光催化剂也可以被用于生命科学,如癌症的治疗[10]。银基的表面等离子体光催化剂可以在癌细胞内高效率地产生大量活性氧物质(ROS),从而产生一定量的细胞毒性引导癌细胞的凋亡。
其原理是在催化剂表面独特的局域形成的等离子体共振(LSPR)效应。在入射光的作用下,贵金属颗粒的传导电子可以发生相干振荡,从而产生强烈的光吸收。不仅如此,贵金属和半导体接触形成的肖特基结,可在金属-半导体界面附近形成内建电场,迫使光生电子和空穴向不同方向移动,减少它们的原位复合,从而提高电荷分离效率[19-21]。
本课题组主要开展了以银基、卤化银(Ag/AgX)系列,金(Au)、铂(Pt)贵金属系列和非贵金属如氧化钨(WO3)的表面等离子体光催化剂的研究工作。
1.1 表面等离子体光催化材料的光催化原理
自20世纪70年代初首次报道在TiO2电极上光解水以来,TiO2因其环境友好、成本低、光化学稳定性好而成为研究最多的半导体光催化剂。然而,TiO2的宽禁带(金红石的禁带宽度为3.0 eV,锐钛矿的禁带宽度为3.2 eV)使其仅对紫外(UV)光区响应[22-23]。紫外光占太阳光的能量不到5%,而可见光和红外光占太阳光能量的90%以上。因此,提高可见光的利用率成为了解决能源问题的当务之急。

图1 可见光照射下表面等离子体催化剂产生H2可能机制的示意图[24]Fig.1 Schematic illustration of possible mechanism of plasmonic H2 generation under visible light irradiation[24]
如图1所示,以Au纳米颗粒附着在TiO2表面为例,通过改变表面等离子体光催化剂中贵金属的大小、形状和周围介质可以实现拓宽表面等离子体光催化剂的光吸收范围,实现从紫外到红外光的吸收,提高体系对光的利用率[24]。其次,贵金属和半导体接触形成的肖特基结(见图1)可以将光生电子和空穴有效地分离,减少它们的原位复合,从而提高光催化剂的效率[19-21]。最后,在半导体表面沉积的贵金属颗粒(见图1)还可以作为电子陷阱和反应的活性位点促进催化反应的进行。这些因素催生了一种新型高效可见光光催化剂,即贵金属颗粒沉积在合适的半导体(如AgCl、AgBr)形成的金属-半导体复合光催化剂。一般而言,可见光表面等离子体光催化剂是一种由贵金属颗粒和半导体组成的复合光催化剂,但最近其他材料如碳、氧化石墨烯(GO)、石墨烯和氧化物绝缘体也被用作载体来形成表面等离子体光催化剂。下文将以银基表面等离子体光催化剂、其他贵金属(如金、铂等)-半导体表面等离子体光催化剂和非贵金属氧化物表面等离子体光催化剂为主要内容做简要总结。
1.2 银基可见光表面等离子体光催化剂
自可见光表面等离子体光催化剂Ag@AgCl被报道以来,大量新的可见光表面等离子体光催化剂被合成[25]。本课题组首次设计并合成了一系列银基表面等离子体光催化剂,在可见光下对有机污染物的降解、癌细胞的灭活以及有毒重金属离子的还原等方面均表现出优异的光催化活性。
银(Ag)是表面等离子体中最重要的材料之一,在可见光到近红外区域有表面等离子体共振效应。与金(Au)、铜(Cu)和铝(Al)等其他金属材料相比,Ag有很多优势。首先,Ag和Au都具有较高的化学稳定性(抗氧化能力强)和较高的自由电子密度(约5.9×1022cm-3),这使得它们在实际使用环境下的稳定能力和对太阳光的有效利用方面优于其他两种材料。然而,Ag(约26.68美元/盎司)比Au(约1 734.78美元/盎司)便宜得多。银纳米颗粒的表面等离子体共振效应是由带内激发对可见光的吸收产生的,它可以根据其形状、大小和周围介质进行调节。
本课题组成功地合成了可见光表面等离子体Ag@AgCl光催化剂[25]。利用离子交换和光诱导的化学还原使银纳米颗粒原位生成在卤化银纳米颗粒表面上,从而合成出Ag@AgX(X=Cl、Br、I)表面等离子体光催化剂[26-27]。如图2所示,在Ag/AgX(X=Cl、Br、I)表面等离子体光催化剂中,来自卤化银粒子的激发态电子与来自银纳米颗粒的等离子体耦合可以增强表面等离子体共振效应,促进光吸收[28]。这使得所制备的Ag@AgCl等离子体光催化剂对甲基橙(MO)染料分解的光催化活性很高,比氮掺杂TiO2(N-TiO2)的光催化活性高出8倍。与此同时,光动力学治疗的试验表明,Ag@AgCl作为一种高效、稳定的表面等离子体光催化剂在可见光下可以有效地杀死80%以上的癌细胞并产生极少的暗生物毒性。

图2 表面等离子体光催化剂Ag/AgX(X=Cl、Br、I)在可见光照射下的自稳定过程示意图[28]Fig.2 Schematic diagrams showing self-stabilizing process of plasmonic Ag/AgX(X=Cl, Br, I) photocatalysts under visible-light irradiation[28]
银与其他宽禁带半导体的复合可以提高其光催化活性。如TiO2(3.20 eV)和ZnO (3.37 eV),可作为另一类被探索使用银纳米颗粒作为载体的重要半导体。银纳米颗粒作为电子储存器,可以提高接触型TiO2的光催化活性。Awazu等[29]将TiO2薄膜沉积在一层SiO2壳层覆盖的Ag纳米颗粒上以防止其氧化,从而制备出Ag@SiO2-TiO2薄膜。由于银纳米粒子的表面等离子体共振效应,样品在近紫外区表现出强烈的吸收(410 nm),与纯TiO2相比,其对亚甲基蓝(MB)染料的分解速率提高了7倍。
除此以外,在银基半导体,Ag的4d轨道通常与O的2p轨道杂化形成新的价带(VB)。通过这种方式,银基半导体具有窄的带隙,增加了对可见光的有效利用。如Ag3PO4(Eg=2.43 eV)、Ag3VO4(2.5 eV)、Ag2O(1.3 eV)、AgAlO2(2.81 eV)、Ag6Si2O7(1.58 eV)等多种银基半导体在可见光下表现出优异的光催化活性[30-33]。
1.3 金、铂、钯等贵金属表面等离子体光催化剂
金、铂、钯贵金属纳米颗粒均具有表面等离子体共振效应,可以通过调控金、铂、钯的大小、形貌以及所处环境来调控光催化剂对可见光的吸收,因此由金、铂、钯构成的表面等离子体光催化剂也受到了人们的广泛关注。在表面等离子体光催化的纳米结构中,金纳米棒因其具有广泛可调的纵横比和宽的可见光吸收范围,作为催化剂载体而被广泛应用。
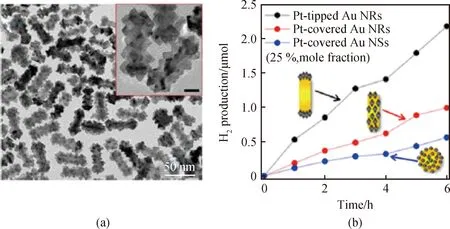
图3 (a)Pt覆盖的Au NRs TEM照片;(b)在可见光照射下(460<λ<820 nm), Pt修饰Au纳米颗粒(0.188 mg)在水-甲醇(体积分数20%)悬浮液中H2演化的时间过程[34]Fig.3 (a) TEM images of Pt-covered Au NRs; (b) time course of H2 evolution from water-bmethanol (20%) suspensions of Pt-modified Au nanoparticles (0.188 mg) under visible-light irradiation (460<λ<820 nm)[34]
如图3所示,本课题组利用十六烷基三甲基溴化铵(CTAB)和5-溴水杨酸组成的二元表面活性剂,建立了铂纳米颗粒在金纳米棒(NRs)上各向异性过度生长的有效途径[34]。通过改变侧面涂层表面活性剂,成功地合成了铂完全包覆的金纳米棒样品。相比于纯金纳米球(NSs),铂完全包覆的金纳米棒呈现出更高的产氢速率。
在此研究的基础上,本课题组又基于表面等离子体双金属纳米结构,利用太阳能在低温下可有效驱动传统的催化反应,开发了钯(Pd)修饰的金纳米棒材料,它可以同时作为光吸收剂和催化活性位点,即使在低于室温(5 ℃)的情况下,也能表现出高效的表面等离子体增强催化甲酸脱氢过程[35]。
1.4 非贵金属表面等离子光催化剂
除贵金属纳米颗粒外,当自由载流子密度达到一定阈值时,掺杂半导体也能显示表面等离子体共振吸收效应(见图4)[36]。基于贵金属的氢溢流效应,本课题组利用氢掺杂来提高半导体的载流子浓度,在低温及常压下实现氢在金属氧化物中的重度掺杂,生成了钼青铜HxMoO3及钨青铜HxWO3。通过理论计算,掺杂进MoO3和WO3中的氢原子能够提供大量的非局域化电子,使得生成的HxMoO3和HxWO3的自由电子密度大幅提高,在可见光区域显示出强烈的表面等离子体共振吸收。随后研究发现,用同样方法生成的半导体钒青铜HxV2O5材料,则由于电子被束缚而未呈现表面等离子体共振效应。该工作从实验和理论两方面证明了氢掺杂效应对于材料载流子浓度的影响,拓宽了具有可见光响应的表面等离子体材料范围。

图4 (a)随氢含量变化的钼青铜HxMoO3的紫外-可见-近红外漫反射光谱;(b)HxMoO3费米能级附近的三维可视化电荷密度分布图[36]Fig.4 (a) UV-Vis-NIR diffuse reflectance spectra of HxMoO3 with hydrogen content variation; (b) three-dimensionalvisualization of electronic charge density distribution around the Fermi level for HxMoO3[36]
2 红外光催化材料
众所周知,在太阳光谱中紫外光、可见光和红外光能量分别占5%、43%和52%[3]。虽然,人们通过各种方式将半导体光催化材料的光响应范围拓展到了整个紫外和可见光区域,但是占太阳光能量一半的红外光能量却没有得到有效地利用。光吸收作为光催化过程的第一步,光催化材料捕获的光子能量多少直接影响光催化反应的能量转换效率的大小。因此,发展能够吸收和利用红外光能量的光催化材料具有重要意义。
2.1 新型上转换红外光催化材料
目前,人们虽然已经探索并发现了一些能够在近红外光照射下具有光催化活性的材料,如:H掺杂的黑色TiO2、氧缺陷型Bi2O3-x红外光催化材料等[37-38]。但是这些材料的红外光催化活性以及光催化氧化还原能力相对于紫外和可见光响应的光催化材料具有较大差距。其根本原因在于,基于传统半导体材料光电效应的光催化材料,其光生电子和空穴的还原和氧化能力受到半导体材料本身能带位置和禁带宽度的限制。因此,对于能够吸收红外光的半导体材料,由于其禁带宽度较低,通常能够吸收近红外光的半导体材料,其光生电子和空穴的氧化还原能力较弱。而近期,人们发现将上转换材料同半导体材料复合形成的上转换光催化材料可较好地解决这一问题。由于上转换材料可吸收红外光而发射出具有更高能量密度的紫外光或可见光,因此当半导体材料同上转换发光材料复合后,可通过吸收上转换材料发出的紫外光或可见光产生红外光催化活性,并保持较高的氧化还原能力,成为吸收和利用红外光的一种有效途径[39-41]。
虽然,上转换红外光催化材料可以在不降低光催化材料氧化还原能力的前提下实现红外光催化。然而,由于上转换光催化材料中近红外光吸收依赖于稀土离子的掺杂引起的离子吸收(如:Yb3+、Er3+等),不同于半导体材料的能带吸收,上转换材料通常对于红外光的吸收强度较弱,并且上转换发光的能量转换效率较低,导致上转换红外光催化材料的整体光催化效率较低。为解决这一问题,能否通过具有较大红外吸收截面的稀土离子作为媒介(如:Nd3+),从而增强传统上转换红外光催化材料的红外光光催化活性。基于这一设想,研究人员设计并制备了NaYF4∶Yb3+,Tm3+@NaYF4∶Yb3+,Nd3+@TiO2(Tm@Nd@TiO2)核壳结构红外上转换光催化材料[42]。如图5所示,Tm@Nd@TiO2复合材料由三层核壳结构组成,最内层为NaYF4∶Yb3+,Tm3+,中间层为NaYF4∶Yb3+,Nd3+,最外层为TiO2。在该复合材料中,由于Nd3+相对于Yb3+具有更大的红外光吸收界面,因此具有更强的红外光吸收能力。并且,通过构建Nd3+到Yb3+到Tm3+的能量传输通道,使得Tm@Nd@TiO2核壳结构复合材料的上转换发光得到极大增强,并且复合材料的界面荧光淬灭得到有效抑制,从而大幅提高了复合材料的上转换发光效率。由于Tm@Nd@TiO2复合材料上转换发光性能得到有效提升,其在光催化降解有机污染物过程中的光催化效率得到显著提升。相对于NaYF4∶Yb3+,Tm3+@ TiO2(Tm@TiO2),Tm@Nd@TiO2复合材料在980 nm,808 nm和(980+808)nm红外激光照射下降解有机染料的光催化活性分别提高了4.40,5.84和9.83倍(见图6)。并且,Tm@Nd@TiO2复合材料在(980+808)nm红外激光照射下降解乙烯的光催化活性相对于Tm@TiO2提高6.4倍。更重要的是,Tm@Nd@TiO2的红外光催化活性可达到紫外光催化活性同等水平(~2/3),使其具有真正的实用价值。
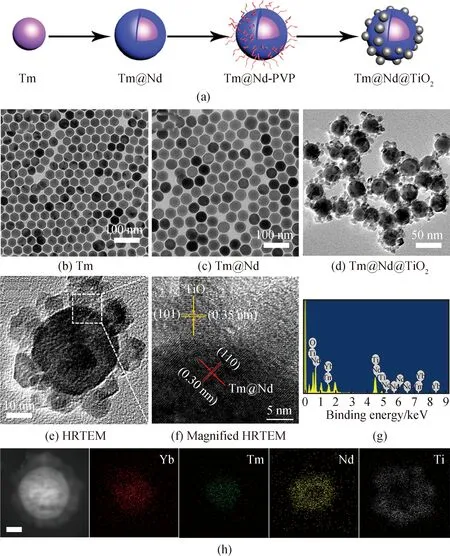
图5 (a)Tm@Nd@TiO2核壳结构的制备过程示意图;(b~f)制备Tm@Nd@TiO2核壳结构复合材料过程中不同步骤中间产物及最终获得的复合材料的TEM和HRTEM照片;(g~h)Tm@Nd@TiO2核壳结构复合材料的EDX和元素分布图[42]Fig.5 (a) Schematic illustration for synthetic procedure of Tm@Nd@TiO2 nanoparticles; (b~f) TEM and HRTEM images of intermediate products and final composite prepared at different steps of Tm@Nd@TiO2 nanoparticles; (g) EDX, and (h) mapping images of a single Tm@Nd@TiO2 nanoparticles[42]
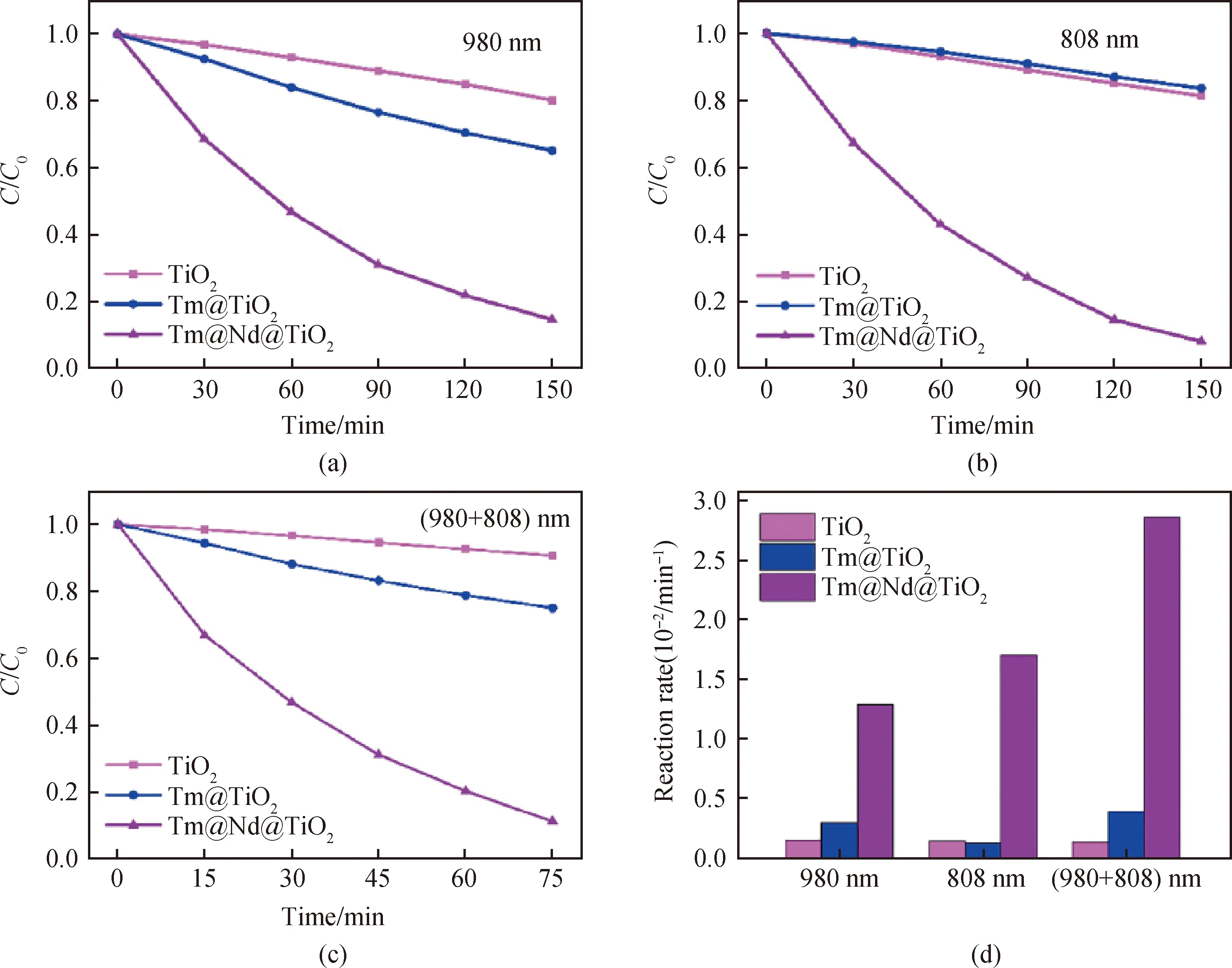
图6 TiO2、Tm@TiO2、Tm@Nd@TiO2分别在(a)980 nm、(b)808 nm、(c)(980+808)nm激光照射下降解RhB染料的降解曲线;(d)TiO2、Tm@TiO2、Tm@Nd@TiO2降解速率的对比曲线[42]Fig.6 Photocatalytic degradation curves of RhB aqueous solution in presence of TiO2, Tm@TiO2, and Tm@Nd@TiO2 under the irradiation of (a) 980 nm laser, (b) 808 nm laser, (c) (980+808) nm laser; (d) comparison of degradation rates in presence of different samples under 980 nm, 808 nm, and (980+808)nm laser irradiation[42]
传统的上转换光催化材料通常由上转换发光材料同半导体材料复合而成,由于复合材料界面的存在,在界面处易产生大量缺陷,这些缺陷将成为上转换发光的荧光淬灭中心和光生载流子的复合中心,因此极大降低上转换发光和光催化反应的能量转换效率。如果能够在单一材料中同时实现上转换发光和光催化两种功能,无疑将解决复合材料界面引起的能量损失,从而进一步提高上转换光催化材料的活性。Bi系半导体材料具有良好的半导体光催化性质,同时Bi3+同稀土离子,如:Yb3+、Tm3+等,具有相似的离子半径。因此,如果将稀土离子掺杂到Bi系半导体材料中,有望在单一半导体材料中同时实现上转换发光和光催化的功能复合。基于上述想法,设计并制备了BiVO4∶Yb3+,Tm3+上转换光催化材料[43]。如图7所示,通常情况下,以Yb3+和Tm3+稀土元素制备的上转换发光材料,如:NaYF4∶Yb3+,Tm3+和LaNbO4∶Yb3+,Tm3+,其上转换发光峰主要位于475 nm、647 nm和704 nm。但当以BiVO4为掺杂介质时,其能带宽度为2.4 eV,可吸收波长小于517 nm的光子能量。因此,BiVO4∶Yb3+, Er3+的上转换发光峰中,475 nm的最强发射峰被BiVO4本身吸收,而仅有647 nm和704 nm两个上转换荧光发射峰。为验证其具有近红外光光催化活性,对其进行光催化性能表征。如图8所示,利用BiVO4∶Yb3+, Er3+作为光催化材料,分别在可见光(>420 nm)和红外光(980 nm激光)照射下降解MB染料。纯的BiVO4及BiVO4∶Yb,Tm材料作为其对比样品。结果表明,在可见光照射下,BiVO4∶Yb3+, Er3+相对于纯BiVO4,在可见光和近红外光照射下均表现出更高的光催化活性。特别是在近红外光照射下,其降解速率常数高达1.6×10-2min-1,在目前已知的上转换光催化材料中处于较高水平[44-48]。该工作为探索同时具有上转换发光和光催化性质的高效上转换光催化材料提供了新的思路。
基于上述思路,为进一步提高红外光催化活性,通过对Bi系半导体材料的能带结构和稀土离子的发射峰位置进行优化,设计制备了具有更高近红外光催化活性的Bi20TiO32∶Yb3+, Er3+上转换光催化材料[49]。由于Bi20TiO32禁带宽度为2.23 eV,相对BiVO4具有更窄的能带宽度,因此,可以吸收发光强度更强的Er3+上转换发光(540 nm)。结果表明,Bi20TiO32∶Yb3+, Er3+在可见光和近红外光照射下降解罗丹明B(RhB)染料的速率可达到0.027 6 min-1,相对于纯的Bi20TiO32降解速率提升近1倍(0.014 27 min-1)。利用Bi系半导体材料作为掺杂基质,不仅可实现上转换发光和光催化的功能复合,在同一种材料中同时实现上转换发光和光催化,还可以通过半导体能带吸收部分上转换发光光谱,实现单色光发射。在光动力学治疗和荧光标记中具有潜在的应用价值。
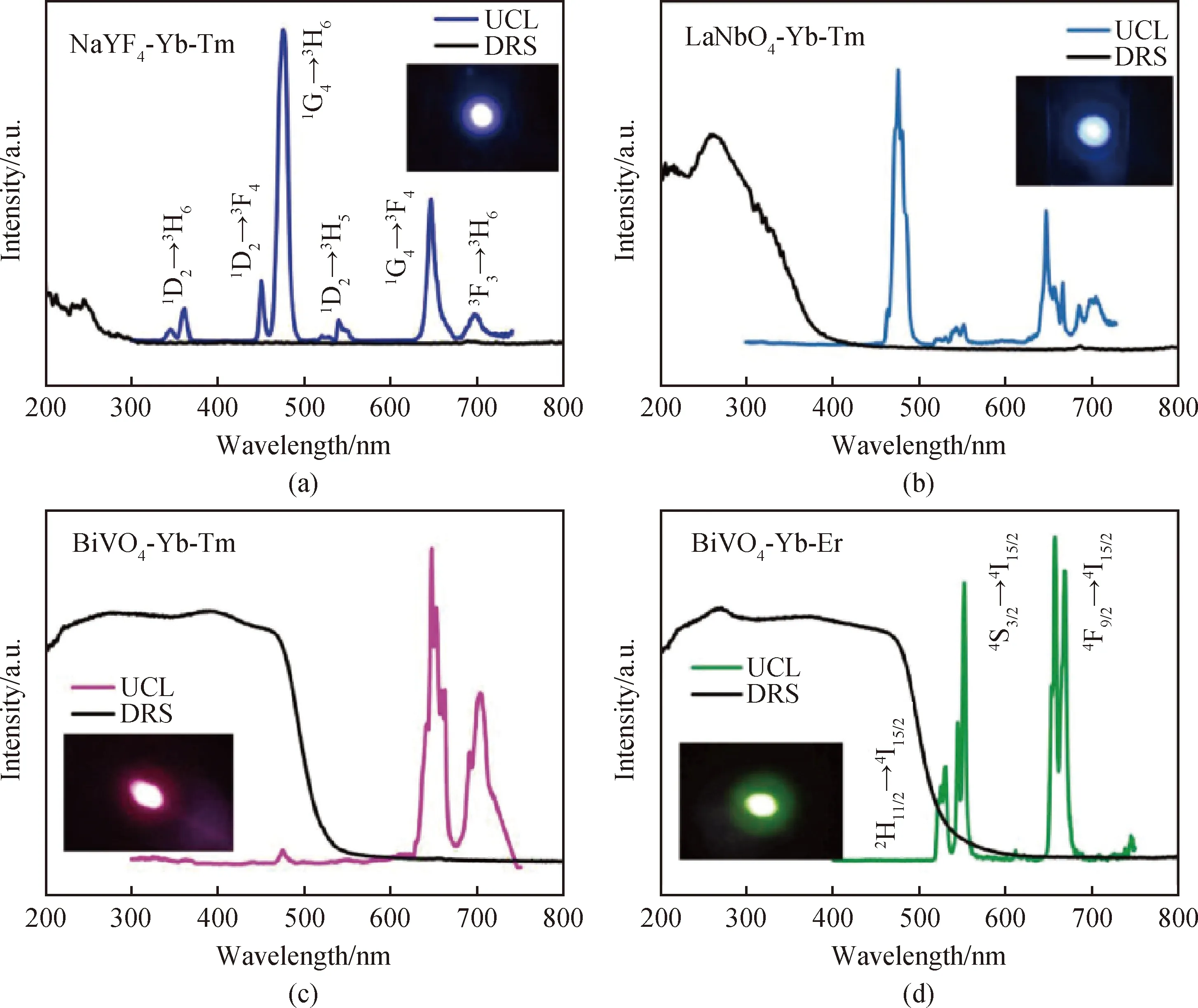
图7 (a)NaYF4∶Yb3+,Tm3+, (b)LaNbO4∶Yb3+,Tm3+, (c)BiVO4∶Yb3+, Tm3+, (d)BiVO4∶Yb3+,Er3+的上转换发光和漫反射光谱,插图显示发光的颜色[43]Fig.7 UCL and DRS spectra of (a) NaYF4∶Yb3+,Tm3+, (b) LaNbO4∶Yb3+,Tm3+, (c) BiVO4∶Yb3+, Tm3+,(d) BiVO4∶ Yb3+, Er3+, each inset shows the color of the luminescence[43]
2.2 基于不同配位离子键电荷转移的新型红外光催化材料
不同于传统的基于半导体缺陷调控的红外光催化材料和基于上转换发光的红外光催化材料,本课题组近期基于对晶体材料的晶体结构和电子结构的分析,发现了一种基于不同配位环境金属离子间电荷转移的新型红外光催化材料[50]。在Cu2(OH)PO4晶体结构中,存在两种不同配位环境的Cu离子,它们分别处于八面体配位(octahedra)和三角双锥配位(trigonal bipyramids)。通过DFT理论计算发现,Cu2(OH)PO4的价带和导带分别由两种不同配位的Cu 3d和O 2p轨道组成(见图9)。当受到光子激发时,电子从Cu2(OH)PO4的最高占据态向四个未被占据能带的跃迁将引发强烈的近红外光吸收。Cu2(OH)PO4的近红外光催化过程主要可归结为光生电子从三角双锥CuO4(OH)中Cu(Ⅱ)位置向与其临近的八面体CuO4(OH)2中Cu(Ⅱ)位置的迁移。如图9所示,由三角双锥CuO4(OH)跃迁至上面三个为占据能带的光生电子,可以很容易地传输到八面体CuO4(OH)2中去。并且一旦光生电子传输到八面体CuO4(OH)2的最低位占据能级,将难以再返回到临近的三角双锥CuO4(OH)中去[50-51],从而使电子和空穴实现有效分离,并促进光催化反应的进行。这一结果表明,基于金属氧化物不同配位环境对材料电子结构的不同贡献,可实现近红外光的吸收和能量转化,可成为设计制备新型红外响应的光催化材料的一种有效途径。该工作发表后,Stephen在美国C&EN上对本工作进行了评论,称本课题组发现的Cu2(OH)PO4是第一个真正具有红外光催化活性的光催化材料,其具有红外光催化活性的关键是其晶体结构中含有两种不同配位环境的Cu离子,并指出该思路为发展太阳能驱动的新型光催化材料提供了一种重要模型[52]。

图9 Cu2(OH)PO4的总态密度(TDOS)和分态密度(PDOS)图,其中黑色曲线表示TDOS图 (a)Cu 3d和O 2p的PDOS曲线;(b)OH基团的O 2p轨道、八面体配位Cu 3d轨道和三角双锥配位的Cu 3d轨道的PDOS曲线;(c)八面体CuO4(OH)2中O原子的O 2p轨道和三角双锥CuO4(OH)中O原子的O 2p轨道的PDOS曲线[50]Fig.9 TDOS and PDOS plots calculated for Cu2(OH)PO4, where TDOS plots are given as a solid black line (a) PDOS plots for total Cu 3d and total O 2p states; (b) PDOS plots for O 2p states of OH groups, the Cu 3d states of octahedral (OCT) Cu sites,and those of trigonal bipyramidal (TBP) Cu sites; (c) PDOS plots for O 2p states of O atoms belonging only to CuO4(OH)2 octahedra and those belonginig only to CuO4(OH) trigonal bipyramids[50]
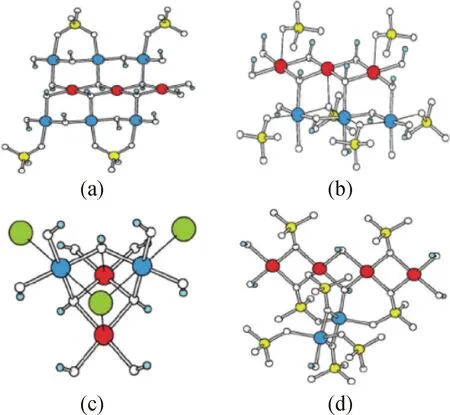
图10 (a)Cu3(OH)4SO4、(b)Cu4(OH)6SO4、(c)Cu2(OH)3Cl、(d)Cu2(OH)PO4的晶体结构示意图[53]Fig.10 Structural features of (a) Cu3(OH)4SO4, (b) Cu4(OH)6SO4, (c) Cu2(OH)3Cl, (d) Cu2(OH)PO4[53]
基于上述工作,进一步对同样具有不同配位Cu离子的三种不同材料,包括:Cu3(OH)4SO4、Cu4(OH)6SO4和Cu2(OH)3Cl进行研究。通过对这三种材料的晶体结构、电子结构和光催化性质进行详细研究发现,Cu3(OH)4SO4、Cu4(OH)6SO4和Cu2(OH)3Cl同Cu2(OH)PO4类似,在晶格中均存在两种不同配位的Cu离子,并通过Cu-O-Cu桥连(见图10)。但只有Cu3(OH)4SO4和Cu4(OH)6SO4表现出类似于Cu2(OH)PO4的近红外光催化活性,具有相似晶体结构和配位环境的Cu2(OH)3Cl却没有任何近红外光催化活性。因此,即使在材料内部存在不同配位的金属离子,并具有电子传输的氧桥存在,材料也不一定具有近红外光催化活性。通过对它们的电子结构进行分析,发现Cu3(OH)4SO4和Cu4(OH)6SO4中的SO4基团同PO4基团类似,都是电子受体基团(见图11)。在电子传输过程中,由于电子受体基团同金属多面体连接,可暂存光生电子,从而促进电子和空穴的传输与分离。而Cu2(OH)3Cl中的Cl为电子给体,无法实现电子的暂时存储,从而无法实现光生电子的传输与分离。因此,在包含通过氧桥连接的不同配位金属离子的金属氧化物中,同时存在电子受体基团是探索和设计具有近红外光催化活性的新型光催化材料的必要条件。该工作为探索和筛选新型红外光催化材料提供了理论依据[53]。

图11 (a)Cu3(OH)4SO4, (b)Cu4(OH)6SO4, (c)Cu2(OH)3Cl, (d)Cu2(OH)PO4的PDOS图[53]Fig.11 PDOS plots calculated for (a) Cu3(OH)4SO4, (b) Cu4(OH)6SO4, (c) Cu2(OH)3Cl, (d) Cu2(OH)PO4[53]
3 极性光催化材料
贵金属负载和半导体复合形成异质结等提高光生电子空穴对的分离效率,具有很大的局限性,像界面电场作用范围小、电场强度较弱,界面处易形成电子-空穴复合中心,使电子-空穴再次复合等。在材料体相建立内建电场,利用极性材料内部自发极化电场作为促进光生载流子分离的驱动力,来提高光生载流子分离的分离效率,是有效解决光生载流子分离效率低,进一步提高光子能量利用效率和改善光催化活性的关键。
3.1 极性基元的定向排布构建内建电场
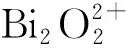
在进一步的研究中,可以发现铋基层状材料Bi2O2[BO2(OH)]也是极性材料。阴离子层[BO2(OH)]2-为极性基团,且在晶体结构中定向排列,在(-2.531 40, 0,-3.358 29)方向上的偶极矩为2.41D。由于导带主要由Bi 6p和O1 2p构成,因此产生于[BO2(OH)]层上的电子e-趋于通过Bi-O2-B键向[Bi2O2]层移动,同时在内建电场的作用下沿电场反方向移动(见图13),最终到达纳米片的边缘通过还原反应被消耗掉;而产生于[Bi2O2]层的电子e-则会直接在电场的作用下向边缘移动,总的来说电子主要向[Bi2O2]层聚集。同样的,由于价带是O 2p构成且O2 2p轨道贡献最大,所以产生于[Bi2O2]层上的空穴h+趋于通过Bi-O2-B键向[BO2(OH)]层移动,同时在内建电场的作用下沿电场线方向移动(见图13),最终到达纳米片的表面并通过氧化反应消耗掉;而产生于[BO2(OH)]层上的空穴h+则会直接在电场的作用下通过O3-B-O2和O3-H-O2等键逐步向晶体表面移动并氧化污染物,因此空穴主要向[BO2(OH)]层聚集。这种光生载流子在纵向上(电子空穴分别在不同层间聚集)和横向上(电子空穴沿内建电场反方向移动)的分离就是Bi2O2[BO2(OH)]具有高光催化活性的最主要原因[6]。
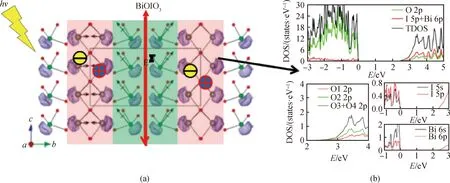
图12 BiOIO3中光生电子空穴在内建电场下的移动方向和相应的态密度图[55]Fig.12 (a) A schematic diagram showing how photo induced electron-hole pairs generated at BiOIO3 pyramids are separated undereffect of the internal polar electric field; (b) total and projected DOS plots calculated for BiOIO3, the energies in the horizontal axes are given in eV[55]
Bi4V2O11是另一种重要的铋基层状材料,阳离子层为[Bi2O2]2+,阴离子层为[VO3.5□0.5]2-。通过计算VO6和BiO4基团的偶极矩,发现该材料在[001]方向上的剩余偶极矩高达91.032D,说明会在[001]方向上产生自发极化电场。态密度计算结果表明,该材料的导带主要由V构成,因此,光生电子将沿着V-O-V层方向传输。价带由O 2p和少量Bi 6s轨道组成,光生空穴将沿着Bi-O-Bi层方向传输,如图14所示。光生电子和空穴分离的方向正好与内建电场的方向是一致的,因此内建电场可促进光生电子和空穴沿着相反的方向快速移动。Bi4V2O11在紫外和全光照射下都表现出良好的光催化产氧活性[57]。

图13 (a)Bi2O2[BO2(OH)]的态密度图;(b)不同氧对能带的贡献;(c)体系中光生电子传输和内建电场方向示意图[56]Fig.13 (a) Total and projected DOS plots of Bi2O2[BO2(OH)]; (b) the different O atoms in Bi2O2[BO2(OH)];(c) a schematic representation showing how the internal polarized field enhances the charge separation and the photocatalytic mechanism of Bi2O2[BO2(OH)][56]
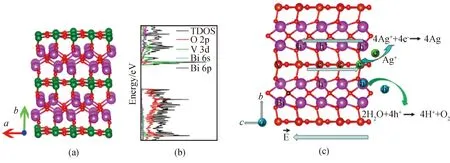
图14 Bi4V2O11的晶体c轴投影结构(a),态密度(b)及光催化过程示意图(c)[57]Fig.14 (a) Crystal structure of Bi4V2O11 projected from the c axis; (b) density of state of Bi4V2O11; (c) schematic illustration of photocatalytic process of Bi4V2O11[57]
除铋基半导体外,几种极性银基材料,也能产生内建电场,促进光生载流子分离。Ag6Si2O7属于单斜相晶体,其中SiO4四面体共角生成Si2O7,12个不同Ag+分为三种配位结构,分别为二配位AgO2、三配位AgO3和四配位AgO4。如前所述,Ag6Si2O7晶体在b轴方向上存在一个剩余偶极矩,可产生自发极化电场,从而促进光生载流子的分离如图15所示。计算其电子结构发现,二配位Ag(a)O2基团对导带底的贡献要比三配位Ag(b)O3基团的大;同时三配位基团对导带底的贡献要比四配位Ag(c)O4的大。所以四配位Ag(c)O4形成的光生电子容易通过Ag(b)-O-Ag(c)桥转移到基团而富集。同样的,三配位Ag(b)O3基团形成的光生电子可以通过Ag(b)-O-Ag(a)桥转移到Ag(a)O2基团而富集[58]。
基于上述思路,进一步发现Ag9(SiO4)2NO3也具有自发极化电场。Ag9(SiO4)2NO3由单独的SiO4四面体基团和NO3三角锥基团与银离子共同配合形成。光催化材料中因为晶格中硝酸根离子的存在使得本来成中心对称的Ag-O-Si框架发生了极化,这种极化使得Ag9(SiO4)2NO3成为一种单斜相的极性晶体。在极性晶体中由于剩余偶极矩的存在会在偶极矩方向存在内建电场。另外由于材料中Ag-Ag键间的d10-d10作用,使得Ag9(SiO4)2NO3具有良好的可见光吸收,表现出1.95 eV的带隙,使得Ag9(SiO4)2NO3成为一种光催化性能优于典型Ag基半导体AgPO4的材料,具体结构及光催化降解MB曲线如图16所示[59]。

图15 (a)Ag6Si2O7的单胞结构,红色代表O,灰色代表Si,粉色代表AgO2中的Ag,黄色代表AgO3中的Ag,绿色代表AgO4中的Ag,绿色圆柱体代表Ag@O键,灰色圆柱体代表Si@O键;(b)晶体结构中Si2O7的排列方式;(c)不等价的Ag中光生电子的传输方向[58]Fig.15 (a) Crystal structure of Ag6Si2O7, showing atoms of a unit cell. Sphere colors: red=O, gray=Si, pink=Ag of AgO2,yellow=Ag of AgO3, green=Ag of AgO4, green cylinder= Ag@O bond, gray cylinder= Si@O bond; (b) the arrangements of Si2O7 units; and (c) directions for photo induced electron transfer between different non-equivalent Ag atoms[58]
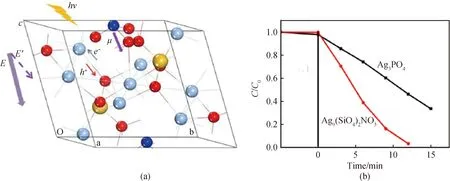
图16 (a)Ag9(SiO4)2NO3晶体结构和极化方向;(b)光催化降解亚甲基蓝曲线[59]Fig.16 (a) Crystal structure and polarization direction of Ag9(SiO4)2NO3; (b) photocatalytic decomposition of MB over Ag9(SiO4)2NO3[59]
在Ag4(GeO4)中,存在GeO4非对称四面体,使得成为Ag4(GeO4)极性晶体。在极性晶体内部存在内建电场,这种内建电场分布于整个晶体中,能够促进光生载流子的分离。因此,虽然Ag4(GeO4)与Ag2O的带隙构成和吸收光谱类似,但前者表现出更高的光催化活性[60]。
3.2 极性分子修饰半导体材料构建表面电场
极性结构自身产生内建电场虽然能够促进光生载流子的有效分离,但是具有极性结构的化合物有限,且大部分极性化合物的光响应范围在紫外甚至深紫外区,严重限制了材料对太阳光的吸收。有机分子结构多样,性能可控,进一步用极性有机小分子对半导体进行表面修饰,得到了具有高效的可见光光催化材料。
利用Bi-S键将具有不同取代基的苯硫酚修饰到甲酸氧铋表面,苯硫酚的对位取代基分别为吸电子基羧基、硝基或卤代基,给电子基胺基或烷基。结果显示,小分子修饰后,材料的吸收带边从368 nm拓展到585 nm。材料的光催化效率和光电流也有明显的提高,两者与取代基团的哈米特常数成线性关系,即小分子的极性越大,光催化效率提高的越多,具体如图17所示。可以推测极性小分子修饰构建了极性界面,促进了光生载流子的分离。这一方法为发展新型可见光光催化材料提供新的思路[61]。

图17 有机小分子修饰无机半导体 (a)拓展光吸收; (b)提高光催化活性和光电流[61]Fig.17 (a) Extended visible light absorption, (b) enhanced photocatalytic activity and photocurrent for organic molecule modified semiconductors[61]
在此基础上,进一步将Bi半导体拓展到BiOX(X=Cl、Br、I)、(BiO)2CO3、和非层状Bi2O3,发现极性苯硫酚的修饰均可以提高上述铋系半导体的光催化活性,证明此方法具有一定的普适性。不仅如此,通过制备不同暴露晶面的 BiOCl 化合物,即{001}晶面和{010}晶面,利用4-羧基苯硫酚(4CBT-H)修饰后,发现4CBT@BiOCl{001}光催化活性提高的程度高于4CBT@BiOCl{010},说明小分子的极性和Bi系层状材料的内建电场存在协同作用[62]。进一步用开尔文探针力显微镜(KPFM)研究小分子修饰前后对表面光电压的影响。结果显示,BiOBr的表面光电压VSP=0.51 V,而对羧基苯硫酚(4CBT)修饰后,4CB@BiOBr的表面光电压提高了50 mV,VSP=0.56 V。这个结果进一步说明小分子修饰产生的极性界面电场促进光生载流子的分离。活性氧物种的测试结果表明,4CBT修饰前,主要活性氧物种是单线态氧1O2,而1O2是激子作用的结果;小分子修饰促进H2O2的生成,而H2O2是电子转移的产物,这进一步证明了极性界面电场的存在,促进了激子到自由电子的转换[63]。
4 光催化材料的微结构调控
材料的物理性能不仅同材料本身的晶体结构密切相关,材料的微结构参数,如:暴露晶面、表面状态、形貌等参数对其物理性能也具有重要影响。随着纳米技术的不断进步,通过对材料的晶面、形貌、表/界面状态进行调控,同样可有效改善材料的光催化性质。近年来,结合纳米材料制备和调控技术,对光催化材料的晶面、形貌、组分等进行调控,进一步提高了其光催化活性,并开发出系列新的材料制备方法、微结构调控手段和增强光催化活性的机理。
4.1 晶面调控
由于晶体的各向异性,其不同晶面表现出具有差异的反应活性,并通过各种作用机制影响着光催化性能。例如,暴露晶面的不同会改变其表面电子能带结构,进而影响光生载流子的生成,晶体取向则会影响半导体内部载流子分离效率。当半导体暴露不同类型晶面时,其表面电子能带结构的差异会导致空间电荷分离,进而在不同晶面分别进行氧化还原反应。因此,设计和制备具有高活性晶面的半导体光催化材料,并通过晶面调控改善其光催化效率,是光催化研究的热点之一[64]。
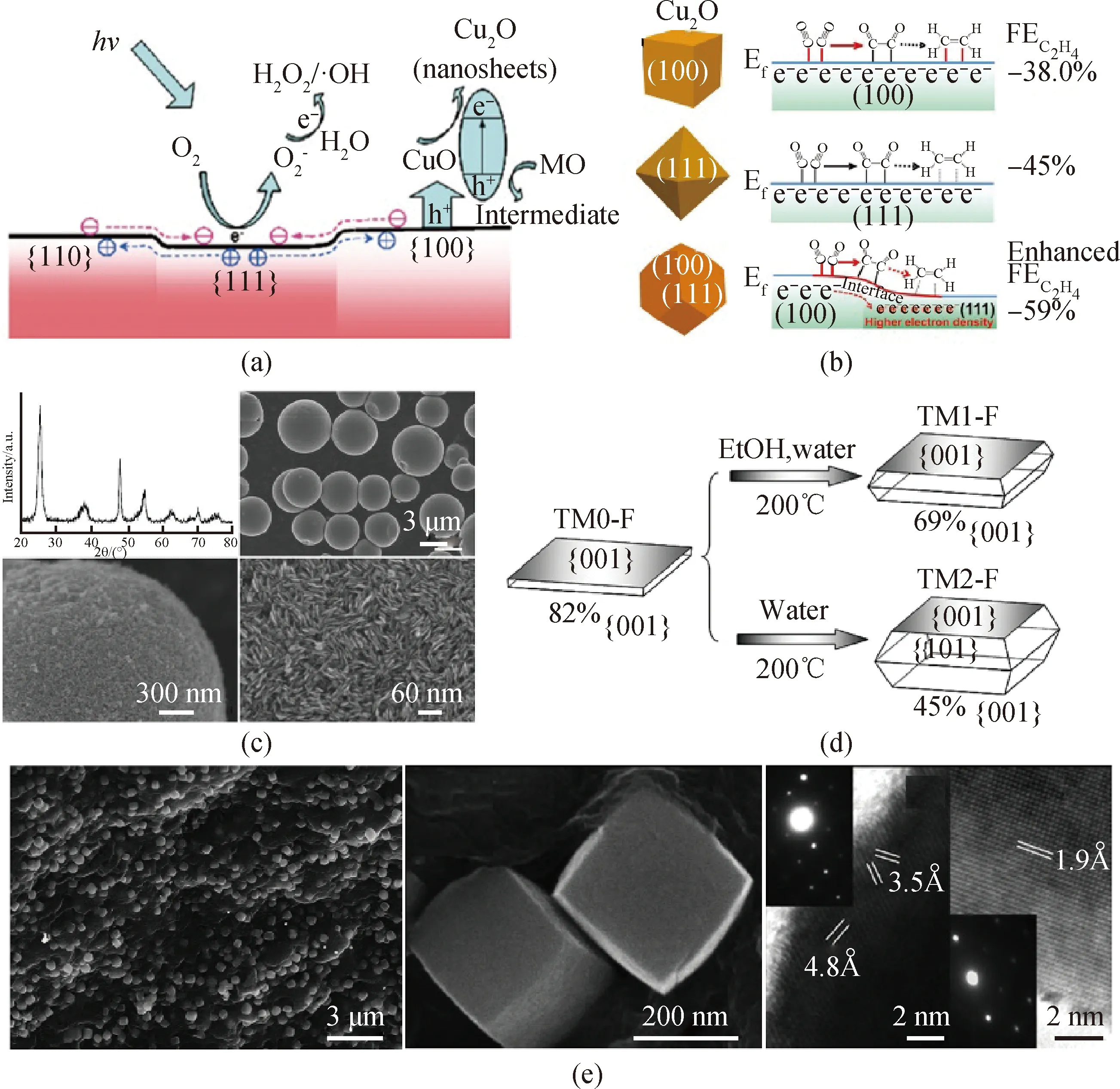
图18 (a)Cu2O纳米片不同晶面之间电荷分离和再生的示意图[65];(b)乙烯在c-Cu2O{100}面、o-Cu2O{111}面和t-Cu2O{100}和{111}面上的生成机制[66];(c)暴露高活性{001}晶面TiO2微球的XRD图谱和SEM照片[67];(d)暴露不同比例晶面TiO2生长示意图[68];(e)石墨烯@TiO2纳米结构的SEM照片[69]Fig.18 (a) Schematic diagram showing charge separation among different crystal faces and regeneration of Cu2O nanosheets[65];(b) formation of C2H4 on the {100} facets of c-Cu2O NPs, {111} facets of o-Cu2O NPs, and {100} and {111} facets of t-Cu2O NPs[66]; (c) XRD pattern and SEM images of TiO2 microspheres exposed with (001) facets[67];(d) growth diagram of TiO2 building blocks in different experimental conditions[68]; (e) SEM images of as-prepared graphene@TiO2 nanocomposites[69]
氧化亚铜(Cu2O)是一种p型半导体,对太阳光谱响应范围较宽,在光催化、气体传感等领域有着广泛的应用前景。本课题组结合晶面调控的策略,制备并研究了Cu2O微晶不同晶面的光催化性质[65]。研究表明暴露{111}面的Cu2O纳米片具有较好的光催化活性,同时还发现在光催化过程中Cu2O{100}和{110}面逐渐消失,并转变成暴露{111}面的Cu2O纳米片,基于试验分析及理论计算,提出了晶面间电荷分离模型(见图18(a)),该模型的提出为促进电荷的有效分离提供了新的思路。半导体表面原子排列决定了反应分子的吸附和活化,进而可调节催化活性和选择性。最近,通过湿化学还原法制备了暴露不同晶面的Cu2O纳米颗粒,研究了其暴露晶面对电催化二氧化碳还原产乙烯选择性和活性的影响,发现乙烯的产率和选择性较大程度上取决于Cu2O所暴露的晶面(见图18(b))[66]。基于试验分析及理论计算,Cu2O{100}和{111}面的协同效应,不仅可促进C-C耦合和乙烯的解吸,而且还可促进产乙烯的多电子动力学过程。
基于晶面调控的思想,采用醇热法在无模板条件下制备了TiO2微球,该微球由暴露高活性{001}面的纳米片组成(见图18(c));由于{001}晶面的高占比(82%)和大比表面积,该材料表现出比商用P25更优异的光催化性能[67]。随后,通过改变二次水热过程中的溶剂,制备出由锐钛矿纳米片或十面体组成的分等级TiO2微球,其中{001}面的占比可由82%调整到45%(见图18(d))[68]。值得注意的是,暴露比例为45%的{001}面的TiO2微球显示出最优异的光反应活性;基于理论计算和试验分析,提出了{001}和{101}晶面间电荷转移模型,晶面间协同效应是光催化活性的决定性因素,该效应可有效促进光生载流子分离。晶面间电荷转移模型的提出为光催化活性的改善提供了一种新思路。此外,本课题组合成了暴露晶面可控的石墨烯@TiO2复合材料(见图18(e)),由于高活性晶面的存在和石墨烯诱导的纳米晶高分散性,该复合材料对产氢和染料降解均表现出增强的光催化活性[69]。基于相同的思路,随后通过溶胶-凝胶法合成了暴露{001}晶面的层状钙钛矿型卤化Bi4NbO8Cl,其在可见光下降解有机染料也表现出优异的性能[70]。
为了获得暴露特定晶面的高效光催化材料,控制其晶体生长过程中的生长取向是一条有效途径。本课题组利用动力学控制原理调控得到暴露不同晶面的Ag2O纳米晶体(见图19(a))[71]。基于理论计算,发现Ag2O{100}面具有比其他晶面更高的表面能,沿<100>方向的空穴和电子的有效质量的差异也是最大的,进而有利于电子空穴对的分离;此外,{100}面具有更合适的氧化还原电势。该结果解释了晶体结构与电子结构对光催化性能影响的本质,对深入理解光催化性能与晶体结构之间的关系具有重要意义。基于同样思路,利用湿化学氧化法合成了AgCl凹面立方体,其中AgCl立方晶种沿<111>和<110>方向优先过度生长,从而获得了凹面结构(见图19(b))[72]。与暴露{001}面的立方体相比,暴露有高折射率面的AgCl凹面立方体在光催化产氧中具有更高的活性。

图19 (a)不同暴露面的Ag2O微米晶的SEM照片[71];(b)凹面AgCl的生长机理图[72];(c)剥落的<100>取向的TaON正面和背面以及(012)LiTaO3衬底的照片(左);剥落的TaON薄膜正面及背面(中)和烧结的TaON薄膜正面及背面光电流图(右)[73]Fig.19 (a) SEM images of Ag2O microcrystals synthesized with various morphologies[71]; (b) growth mechanism of concave AgCl[72]; (c) photographs taken for the front- and back-side surfaces of exfoliated <100>-oriented TaON as well as the (012) LiTaO3 substrate (left); J-V bias relationships determined for front and back sides of the asprepared exfoliated TaON film (middle) and annealed exfoliated TaON film (right)[73]
相较于粉体光催化材料,对光电极进行晶面调控以实现高效的光电水分解更具挑战性。氧氮化钽(TaON)具有约2.4 eV的带隙,适合吸收可见光,已成为一种有前途的光阳极材料。但其较差的粒子间电荷传输特性和较高的电荷复合率限制了其在光电水分解中的应用。为了增强TaON光阳极的水分解性能,通过在NH3/CCl4混合气体下氮化LiTaO3{012}单晶来制备<100>取向的TaON纳米多孔膜[73]。这种定向的TaON多孔纳米结构促进了光生电子在整个膜上的有效转移,从而增强了光生电子-空穴对在膜表面的分离,并表现出优异的光电性能(见图19(c))。
光催化材料的晶面调控已成为提高光催化性能的重要研究方向。通过改进晶面控制和微结构调控的合成技术,并借助先进的表征技术来研究界面处的电荷传输动力学,对于半导体光催化材料性能的提升具有重要指导意义。
4.2 光催化材料的形貌调控
材料的形貌调控对材料的催化活性至关重要。在结构与性能的相互关系方面,结合纳米材料的制备技术,本课题组对部分微纳晶体材料的微结构进行调控,如:尺寸、晶面、物相等,并研究了这些参数与材料光催化性能之间的关系。
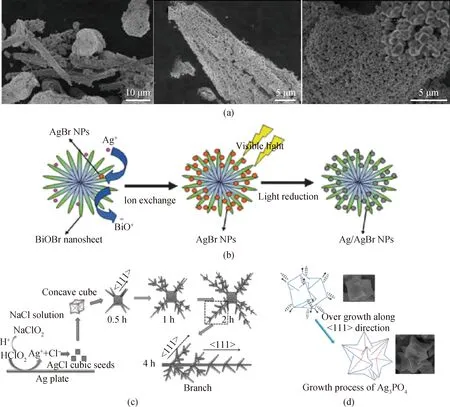
图20 (a)不同形貌的Ag@AgCl表面等离子体光催化材料[74];(b)Ag/AgBr/BiOBr 纳米异质材料的合成示意图和形貌[75];(c)沿<111>方向生长的三维枝状AgCl微米晶体[76];(d)电化学合成凹面Ag3PO4微晶的生长机理示意图[77]Fig.20 (a) Different morphology Ag@AgCl surface plasma photocatalytic materials and photocatalytic curves of degrading MO[74]; (b) synthetic schematic diagram and morphology of Ag/AgBr/BiOBr nano heterogeneous materials[75];(c) three-dimensional dendritic AgCl microcrystals grown along <111> direction[76]; (d) schematic diagram of growthmechanism of electrochemically synthesized concave Ag3PO4 crystallites[77]
4.2.1 银基纳米晶光催化材料的形貌调控
基于Ag@AgCl表面等离子体光催化材料的研究基础,对该类材料进行了微结构调控(见图20(a));通过简单的离子交换过程(使用不同的前体)和光诱导的化学还原反应,制备出一系列具有不同形貌的表面等离子体光催化材料,如:微柱、微球和空心球,并研究了其光催化活性同材料微结构之间的关系[74]。其中,具有空心球结构的Ag@AgCl样品表现出最高的光催化活性,这归因于其具有较多的反应活性位点。在材料复合方面,通过原位离子交换反应合成了具有可见光响应的Ag/AgBr/BiOBr高效复合光催化材料(见图20(b)),该材料集成了Ag/AgBr表面等离子体和AgBr/BiOBr半导体复合光催化材料的协同效应,具有非常好的光催化抑菌和染料降解效果[75]。另外,根据在AgCl晶体生长方面的研究基础,采用湿化学氧化法,通过改变AgCl生长过程中Ag+和Cl-的比例,控制立方晶核在<111>方向上定向生长,得到了分等级三维枝状AgCl微米晶体(见图20(c)),由于其表面暴露了大量的活性面,该材料在光催化裂解水产氧中具有很高的活性[76]。随后,利用电化学方法合成了凹面Ag3PO4纳米晶(见图20(d)),由于凹面晶体的表面由高指数面构成,高指数面存在大量的原子台阶,为催化反应提供了大量的活性位点,进而表现出较高的光催化活性[77]。
4.2.2 TiO2基光催化材料的形貌调控
分等级结构纳米材料是材料科学领域被广泛关注的一类新型纳米材料。在光催化领域,具有分等级结构的光催化材料由于其比表面积大、活性位点多以及易分离回收等特性,通常表现出更加卓越的催化性能。针对二氧化钛这一重要的光催化材料,利用碱热法合成出了具有分等级结构的TiO2纳米管微球,该微球是由TiO2纳米管交织缠绕而成,如图21(a)所示。

图21 (a)分等级TiO2微球的SEM照片[78];(b)活性面TiO2微管的SEM照片[79];(c)金红石相TiO2分等级微球的SEM照片[80]Fig.21 (a) SEM images of graded TiO2 microspheres[78]; (b) SEM images of active surface TiO2 microtube[79];(c) SEM images of rutile graded TiO2 microspheres[80]
由于该微球具有三维分等级的管状结构及很高的比表面积,其在光催化降解苯酚实验中显示出优异的光催化性能,并有望应用于太阳能电池、催化载体及水处理等领域[78]。随后,通过采用ZrO2纤维作为模板,利用(NH4)2TiF6作为反应原料,通过水热法一步合成了由暴露{001}面微晶组成的TiO2微管(见图21(b)),并提出了其晶体生长机制:在(NH4)2TiF6分解产生的F-的作用下,TiO2高活性{001}面得以显露,并附着在ZrO2纤维表面,而ZrO2纤维在反应过程中所产生HF腐蚀下被逐渐溶解,从而形成由{001}活性面TiO2微晶组装而成的TiO2微管[79]。此外,采用一步水热法制备了金红石相分等级TiO2微球(见图21(c)),该微球由暴露{110}面的金红石TiO2纳米棒组成,由于其增强的结晶质量、较高的{110}面暴露比例以及更大的空隙率,该材料显示出优异的光催化性能[80]。
4.2.3 Bi基光催化材料的微结构调控
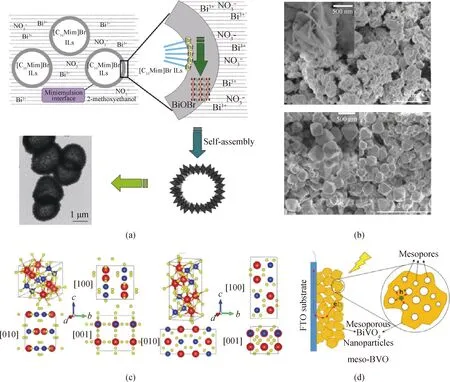
图22 (a)BiOBr空心微球在微乳液界面形成示意图[81];(b)Bi2WO6纳米材料在不同pH条件下的形貌[82];(c)单斜白钨矿相和四方锆石相的晶体结构[83];(d)介孔BiVO4电子空穴传输示意图[84]Fig.22 (a) Schematic diagram of formation of BiOBr hollow microspheres at microemulsion interface[81]; (b) morphology of Bi2WO6 nanomaterials under different pH conditions[82]; (c) crystal structure of monoclinic scheelite phase and tetragonal zircon phase[83]; (d) schematic diagram of electron hole transport in mesoporous bismuth vanadate[84]
传统制备空心的结构通常需要借助于模板,包括软模板和硬模板。然而模板法步骤冗繁,并且可能会引入杂质。基于乙二醇甲醚和含溴离子液的有限互溶度,利用其搅拌产生的微乳液作为反应室,通过一锅法合成了BiOBr空心微球(见图22(a))。其中含溴离子液不仅可作为溴源,而且可用来产生乳液,所形成的含溴微乳液在BiOBr中空心微球结构的反应和自组装过程中起到重要调节作用。BiOBr空心微球对有机染料的降解和重金属离子的还原具有良好的光催化效果[81]。此外,作为铋基化合物中的一大类,Aurivillius 层状化合物具有结构通式(Bi2O2)[An-1BnO3n+1]。其中,Bi2MO6是最为简单的一类Aurivillius层状化合物,其结构是由(Bi2O2)2+层和(MO6)2-(M=W, Mo)八面体层交替堆垛而成。本课题组研究了pH值对水热制备Bi-W-O微观形貌的影响(见图22(b)),并指出前驱体溶液的酸碱性对生成的Bi2WO6晶相及结晶化程度有很大的关系[82]。可以认为Bi2WO6的层状结构有利于光生电子-空穴在[Bi2O2]2+层和[WO4]2-层间传输,从而降低了相应的载流子重新复合,提高了光催化效率。BiVO4作为光电催化材料的研究热点,具有四种晶体形态。其中,单斜白钨矿相BiVO4在光催化、光电催化等领域已经被广泛研究,但是四方锆石相BiVO4用于太阳能分解水的研究工作较少。近期,本课题组成功制备了四方锆石相BiVO4和单斜白钨矿相BiVO4(见图22(c)),并将两者串联起来构筑光电化学(photoelectrochemical, PEC)PEC无偏压全解水[83]。另外,也利用造孔剂制备了介孔单斜白钨矿相BiVO4,基于其介孔特性进一步促进载流子分离(见图22(d)),改善了其光阳极动力学过程[84]。
5 结语与展望
随着人类社会的不断发展和科技的不断进步,人类社会对能源的消耗日益增大,随之而来的环境污染问题日益严重,目前能源和环境问题已越来越严峻。光催化技术作为一种利用太阳能解决能源和环境的新技术,在未来解决人类社会能源短缺和环境污染问题方面具有极其重要的应用前景。而作为光催化技术核心的光催化材料是实现光催化实际应用的关键。近年来,从晶体学基本原理出发,研究人员基于材料结构与性能的关系,通过对材料的晶体结构、电子结构及微结构参数进行广泛和深入的研究,探索了一些具有创新性的材料设计理论、材料制备方法和微结构调控手段,设计制备了一系列具有宽光谱吸收和高载流子分离效率的新型光催化材料。本文中,基于材料结构与性能的关系,对基于晶体学基本原理的高效光催化材料的设计理论、制备方法和微结构调控手段进行了简要的综述,期望这些结果对探索设计新型高效光催化材料能够提供一些新的启发,从而进一步推动光催化技术的发展及其实际应用。
猜你喜欢
红外技术(2022年11期)2022-11-25
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
中国典型病例大全(2022年7期)2022-04-22
陶瓷学报(2021年5期)2021-11-22
石油化工高等学校学报(2021年3期)2021-07-15
纺织科学研究(2021年1期)2021-03-19
陶瓷学报(2019年6期)2019-10-27
现代工业经济和信息化(2016年8期)2016-05-17
浙江农业科学(2016年11期)2016-05-04
