
Two-dimensional MnN utilized as high-capacity anode for Li-ion batteries∗
2021-05-06 08:55JunpingHu胡军平ZhangyinWang王章寅GenruiZhang张根瑞YuLiu刘宇NingLiu刘宁WeiLi李未JianwenLi李健文ChuyingOuyang欧阳楚英andShengyuanYang杨声远
Chinese Physics B 2021年4期
Junping Hu(胡军平), Zhangyin Wang(王章寅), Genrui Zhang(张根瑞), Yu Liu(刘宇), Ning Liu(刘宁),Wei Li(李未), Jianwen Li(李健文), Chuying Ouyang(欧阳楚英), and Shengyuan A.Yang(杨声远)
1Key Laboratory of Optoelectronic Materials and New Energy Technology,Nanchang Institute of Technology,Nanchang 330099,China
2Department of Physics,Laboratory of Computational Materials Physics,Jiangxi Normal University,Nanchang 330022,China
3Research Laboratory for Quantum Materials,Singapore University of Technology and Design,Singapore 487372,Singapore
Keywords: first-principles calculations,Li-ion batteries,energy storage,physical propertyies
1. Introduction
The Li-ion batteries (LIBs) have been playing a significant role in the commercial energy storage market. The range of applications for different types of LIBs includes portable electronic devices, electric vehicles, and large-scale power grids, and continues to expand rapidly.[1–5]Under this development, a key challenge is to further increase the storage capacity of LIBs. This problem has attracted tremendous research effort, and it involves improving multiple components of the battery design.[6–15]
As an important component of the LIB system,anode materials have received increasing attention in recent years. The traditional anode material is made of graphite. However, its theoretic capacity (∼372 mA·h/g) is not high, and its rate performance is also poor.[16]Thus,there is large room of improvement to enhance the LIB capacity and performance by replacing graphite with some new anode materials.
In the last decade, this direction of research has gained great impetus from the rapid advance of two-dimensional(2D)materials. 2D materials possess several advantages to be used as electrode materials:[17–32](i) 2D materials have large specific surface areas;(ii)they provide atomically sharp and clean interfaces;and(iii)many 2D materials have high electric conductivity. Feature(i)directly boosts the the Li storage capacity. Feature (ii) is beneficial for the Li diffusion on the surface,which is crucial for the battery rate performance.Feature(iii)is also a necessity for any electrode material. In addition,generally speaking,2D materials have relatively small volume changes during lithium ion embedding and de-embedding,which is a requirement for maintaining the structural integrity of the electrode.[33,34]
Because of the above reasons, many 2D materials have been studied for LIB anode applications. In terms of their constituent elements, these 2D materials can be divided into two categories, those containing metal atoms and those not.Generally speaking, there are two approaches to increase the theoretical capacity of anode materials: one is by increasing the amount of embedded lithium, and the other is by minimizing the weight of the anode material. Compared to the materials made of light non-metal elements which are favored by the latter approach,2D materials with metal elements typically have a higher weight, but meanwhile, they also tend to accommodate more Li,and usually they exhibit good electric conductivity. A representative class of such materials is the MXenes, which has been extensively studied for LIB applications both theoretically and experimentally. However,their practical capacity is not satisfactory, possibly because of the undesired surface passivation during the preparation process.
Recently, a new 2D material MnN was proposed and revealed to be a 2D nodal-loop half-metal.[35,36]The material can be synthesized by a spontaneous graphitic conversion from the ultrathin (111)-oriented cubic MnN.[37]It has an out-ofplane ferromagnetic ground state with interesting fully spinpolarized nodal loops.[35]Not considering the magnetic property,2D MnN manifests three features desirable for LIB applications. First,the material is of a single atom thickness,which further minimizes the volume and should facilitate the ion diffusion on its surface. Second, the material is metallic with a good conductivity. Third,both elements Mn and N are rich in nature. The metal element Mn comprises about 0.1% of the earth crust and is the 12thmost abundant element on earth.[38]This is important for lowering the cost.
Motivated by the above considerations, based on firstprinciples calculations, we investigate here the physical and energy storage properties of 2D MnN as an electrode material for LIBs. We find that it satisfies almost all the requirements for a good anode material. Specifically, we demonstrate that MnN is mechanically, dynamically, and thermodynamically stable. The configurations before and after lithium adsorption exhibit good electrical conductivity. The study of Li diffusion on its surface reveals a very low diffusion barrier(∼0.12 eV),indicating excellent rate performance. The calculated average open-circuit voltage of the half-cell at full charge is also very low (∼0.22 V), which facilitates higher operating voltage.Its corresponding theoretical capacity is above 1554 mA·h/g,more than four times that of graphite. In addition, the lattice changes of the material during lithium intercalation are very small (∼1.2%–∼4.8%), which implies good cycling performance. Together,these results reveal that 2D MnN could be a promising anode material for LIBs.
2. Computational details
Our first-principles calculations are based on the density functional theory (DFT) using the plane-wave pseudopotentials[39,40]as implemented in the Vienna ab initio simulation package (VASP).[41,42]The exchange–correlation functional is modeled in the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE)realization.[43]Manganese 3d52s2, nitrogen 1s22s2p2, and lithium 1s22s electrons are deemed as valence electrons in all the following calculations. A cutoff energy of 520 eV is introduced for the plane wave expansion. The Brillouin zone is sampled with 5×5×1 Monkhorst–Pack k-point mesh[44]for the structural optimization, and with 9×9×1 mesh for the electronic structure calculations. The convergence criteria for the total energy and ionic forces are set to be 10−5eV and 10−3eV/˚A,respectively.[33]In our calculations, periodic images of a monolayer are separated by vacuum layers with thickness larger than 18.0 ˚A,so that the artificial interactions between the images are negligible.[33]
The adsorption energy(Ead)[33]for Li atom on the surface is defined as
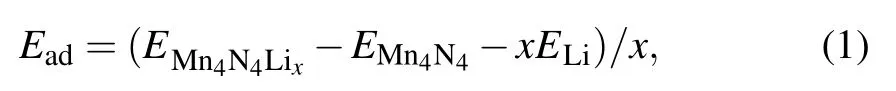
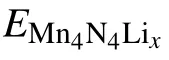
Then the energy difference provides an estimated average open-circuit voltage for a half-cell reaction involving xLi ions

The corresponding theoretic specific capacity could be estimated from the following equation:

where x represents the maximum number of electrons involved in the electrochemical process,F is the Faraday constant with the value of 26789 mA·h/mol,and MMnNis the mass of MnN in g/mol.
3. Results and discussion
3.1. Lattice structure and stability
Before proceeding to investigate its energy storage behavior,we first study the intrinsic crystal structure of 2D MnN in detail. The lattice structure is shown in Fig.1,which belongs to the space group of P-6M2(D3h-1). There is only one bond length in the structure, with the value of 1.928 ˚A. The optimized lattice constants a and b are both equal to 6.679 ˚A.These values agree with previous results.[35]
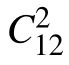
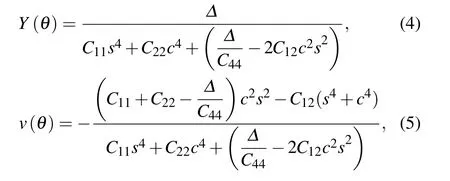
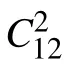

Fig.1. (a)Top view of MnN monolayer,in which the unit cell and all considered adsorption sites are plotted,the θ dependence of(b)Young’s module and(c)Poisson’s ratio for MnN monolayer.
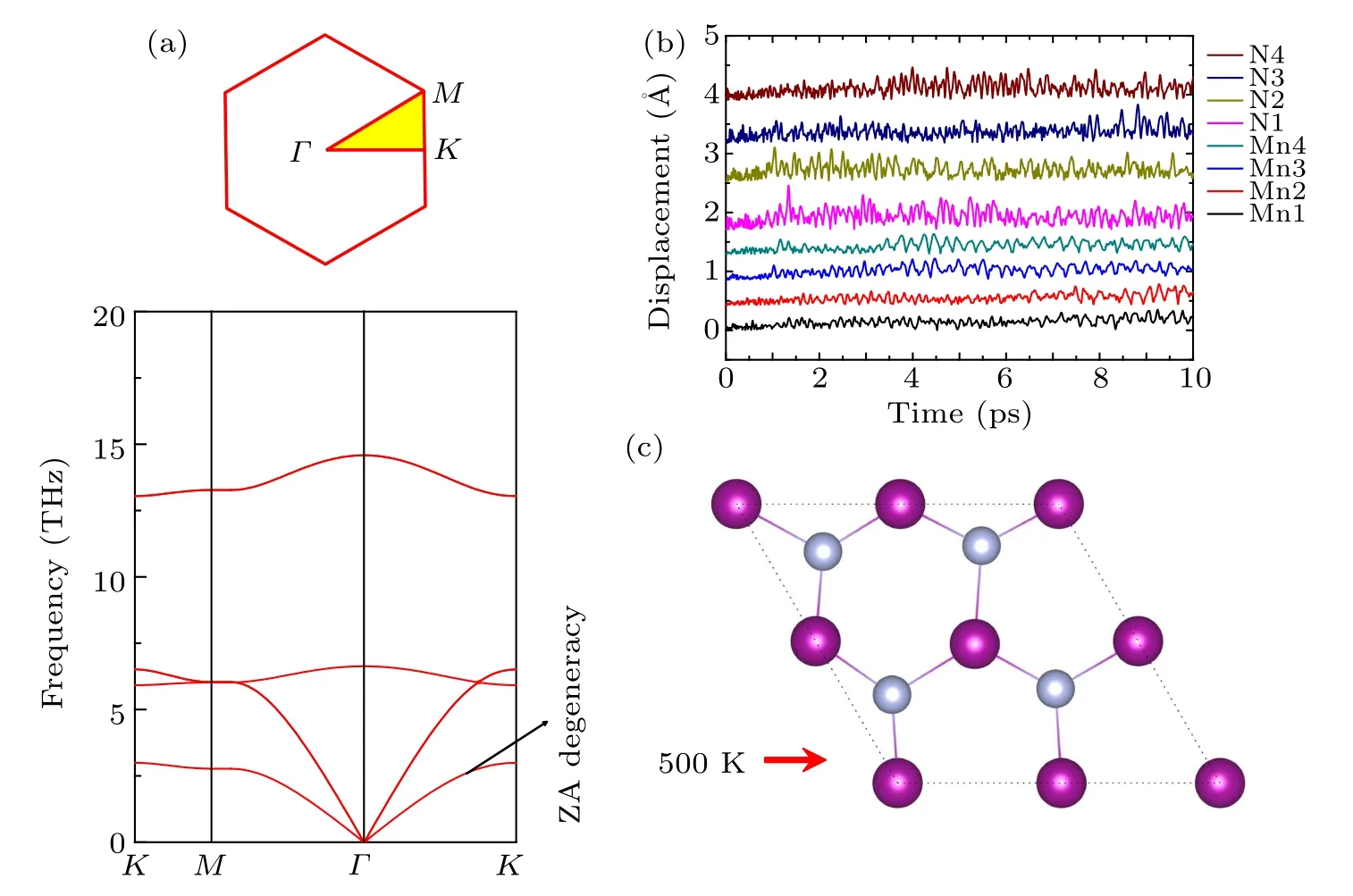
Fig.2. (a) The calculated phonon spectrum and the corresponding Brillouin zone for monolayer MnN. (b) Result from AIMD simulation,including the time-resolved displacement of all atoms in MnN monolayer.(c)The structure of MnN monolayer after 10 ps in AIMD simulation.
As for the dynamic and thermodynamic stability of 2D MnN, it can clearly be seen from the phonon spectrum and the molecular dynamics simulations. As shown in Fig.2(a),there is no imaginary-frequency phonon mode,indicating that the material is dynamically stable. Close to the point, there are two linearly dispersed in-plane acoustic branches, one of which has degeneracy with the out-of-plane acoustic (ZA)branch. We have also conducted the ab initio molecular dynamics(AIMD)simulations. The displacements of the atoms at 500 K over the simulation period of 10 ps are plotted in Fig.2(b),and the snapshot at 10 ps is plotted in Fig.2(c). The results show that the material remains stable. We have also tested the simulation up to 2000 K,and the atomic structure is still kept(see Fig.S1). All these results indicate that 2D MnN is very stable.
3.2. Li adsorption
The Li adsorption energy is a critical indicator to judge whether a material can be used as electrode material or not. In addition,it is required that the material remains a good metal during the adsorption process.
To study Li adsorption on 2D MnN, the most favorable adsorption sites should be identified first. Based on the symmetry, we considered four initial adsorption sites, as shown in Fig.1. These initial positions can be classified into three categories: (1)the hollow site locating above a six-membered ring; (2) the top sites locating on top of Mn or N atoms; (3)the bridge site locating above the midpoint of the Mn–N bond.Moreover,due to the mirror symmetric structure,we symmetrically put two Li atoms on both sides of the 2D layer.
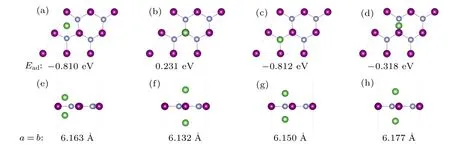
Fig.3. Top view of Li adsorption on(a)hollow site,(b)Mn top site,(c)N top site,and(d)Mn–N bridge site;(e)–(h)the corresponding side view. The atomic color is the same as that in Fig.1.
The adsorption energy is calculated by Eq. (1) to determine which adsorption site Li prefers. The lower the corresponding adsorption energy, the more stable the corresponding configuration. In addition, negative or positive value of the adsorption energy indicates whether Li can spontaneously adsorb or not. The calculated Li adsorption energies for the different sites are presented in Table 1. There,the stability of the adsorption is ranked in the order from high to low,for adsorption at the top site(N),the hollow site,the bridge site,and the top site(Mn).Among them,the most stable adsorption site is the hollow site.

Table 1.Adsorption energies for Li at different adsorption sites on MnN monolayer,and the corresponding adsorption heights,which are defined as the vertical distance from the Li-ion to the center plane. The Li–Mn and Li–N distances are the distances from Li to its nearest neighboring Mn or N atoms.
Next,we utilize the Bader charge analysis to analyze the charge transfer that occurs between Li atoms and the MnN host. For the hollow adsorption site,we find that each Li loses∼0.84e, each Mn atom loses ∼1.14e, and each N atom obtains ∼1.56e. For the top adsorption site (N), we find that each Li loses ∼0.82e,each Mn atom loses ∼1.10e,and each N atom obtains ∼1.51e. In both of these adsorption configurations, the Li atoms become positively charged, while the host is negatively charged.
Then, let us check the metallicity of the material during the Li adsorption. In Fig.4,we plot the calculated local density of states (DOS)[47]of MnN before adsorption and after adsorbing Li at different sites. We find that 2D MnN can keep the metallic property before and after Li adsorption. This satisfies the requirement for a proper LIB electrode material.

Fig.4. The electronic properties: (a)local density of states for pristine MnN monolayer,and(b)–(e)its corresponding four optimized adsorption configurations.
3.3. Li migration on the MnN surface
Besides electronic conductivity,ionic diffusion is another important property. The faster migration of Li ions in the electrode material,the better the rate performance of its corresponding lithium-ion battery.To investigate the Li diffusion on 2D MnN,we use a 4×4 supercell containing 16 Mn and 16 N for calculation. The climbing-image nudged elastic band(Cl-NEB) method[48]is used. This method determines not only the value of the diffusion potential barrier,but also the saddle point. Based on the previous adsorption energy calculations,we select the two nearest neighboring and most stable adsorption configurations as the initial and final states, respectively.Following from the symmetry,we consider the diffusion process along three paths,as shown in Fig.5(a). Path-I and path-III indicate that Li migrates through the top site of Mn and the hollow site,respectively,in the process of diffusion. Path-II represents that Li migrates directly from the initial to final positions.
For all three diffusion processes, the calculated energy barrier diagrams are shown in Fig.5. After Cl-NEB relaxation, the optimized pathways of path-I and path-III are almost the same as the initial ones,but path-II relaxes into path-III.The lowest barrier occurs through path-III,with a value of 0.12 eV.Typically,a diffusion barrier below 0.6 eV is considered acceptable. In comparison, the value of 0.12 eV is very low, which indicates that excellent rate performance can be expected for 2D MnN.
Fig.5. (a) The considered Li migration pathways and (b) the corresponding diffusion-barrier profile along the three pathways.
3.4. Average open-circuit voltage and theoretic storage capacity
The average open-circuit voltage (OCV) and the storage capacity are another two important properties for LIBs. To investigate these two,based on our discussion of the adsorption energies of Li at various adsorption sites in Subsection 3.2,we will progressively raise the amount of Li atoms symmetrically adsorbed on both sides of 2D MnN.In the calculation,we continue to adopt the 2×2 supercell. By launching the half-cell reaction model as follows:
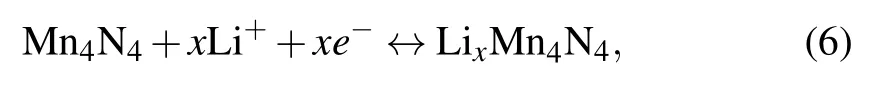
the average OCV can be obtained as an approximation from the energy difference according to Eq.(2).
Based on the adsorption energy of different sites,the order is to cover the top site(N)first as the first layer(symmetrically on both sides),followed by the hollow site(Mn)as the second layer, and finally the top site (Mn) as the third layer.This is displayed in Fig.6. To evaluate the coupling between the Li layer and the host material, we compute the average adsorption energy of Li atoms in each sequential layer(Eav),which is defined as[49]


The corresponding average adsorption energies are calculated to be −0.343 eV,−0.106 eV,and −0.039 eV when Li is adsorbed in the first, second, and third layers in sequence,respectively. The Eavvalues for both the first and the second layers are below −0.1 eV,indicating that at least two layers of Li can be adsorbed on each side of 2D MnN(totally four layers of Li for both sides). Meanwhile,we note that the Eavvalue is still negative even for the third layer,implying that there may also form a third Li layer. However, this negative Eavvalue is small. As a conservative estimation, we do not take the third layer into account. Then,each 2×2 MnN supercell can accommodate up to 16 Li atoms. The corresponding chemical stoichiometry is Li16Mn4N4(or Li4MnN).The theoretical specific capacity is 1554 mA·h/g according to Eq.(3), which is four times that of graphite (∼372 mA·h/g for LiC6) and∼16 times that of pure graphene(∼93 mA·h/g for LiC24).[33]If we further include the third layer on both sides of 2D MnN,the estimated capacity can even reach 2331 mA·h/g.

Fig.6. Side view of the considered multilayer model: (a) Li8Mn4N4,(b)Li16Mn4N4,(c)Li24Mn4N4,and their corresponding side view: (d)Li8Mn4N4,(e)Li16Mn4N4,(f)Li24Mn4N4.
The capacity of 2D MnN is remarkably high. To illustrate this, we compare this value with those of several other 2D anode materials proposed so far. In Fig.7, we compare MnN’s performance separately with the 2D materials containing metal elements and the 2D materials without metal elements. One can observe that MnN’s capacity outperforms most members in each figure. Particularly, comparing with the materials containing metal elements, MnN is the second best among the materials reported to date. Its value is only lower than that of Mg3N2. We need to point out that the value 1554 mA·h/g of 2D MnN used for comparison here is a conservative estimation. The value of 2331 mA·h/g when including the third Li layer would outperform all previous reported values.
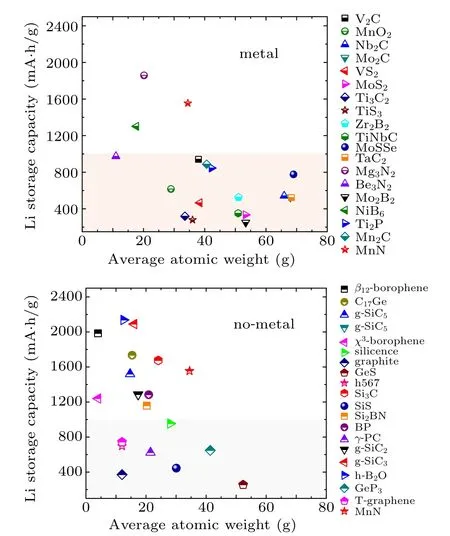
Fig.7. Comparison of the theoretical Li storage capacity values between MnN monolayer and other 2D anode materials (plus graphite):(a)Host materials only contains non-metallic elements,(b)Host materials contain metal elements.
Finally, by using Eq. (2), the calculated average OCV is 0.22 V, which is quite low, desired for anode materials. And the lattice parameters change from 1.2%to 4.8%during intercalation,indicating that 2D MnN should also enjoy great cycle performance.
4. Conclusion
By using first-principles calculations,we predict that 2D MnN can be a promising high-capacity LIB anode material.We demonstrate that the material has excellent mechanically,dynamically, and thermodynamically stability, good electrical conductivity before and after Li adsorption, very low diffusion barrier (∼0.12 eV), low average open circuit voltage(∼0.22 V), and small lattice expansion (∼1.2% to ∼4.8%)during Li intercalation. These properties ensure its superior rate,cycle,and stability performances. More importantly,we find that its theoretical capacity can be above 1554 mA·h/g(Li4MnN), which is four times that of graphite. The high capacity is connected with the facts that (1) the single-atomthickness allows the exposure of more adsorption sites;and(2)the adsorption energies of Li are very close to each other for sites on the top of the hollow site and the N site. Our work not only predicts a promising anode material for LIBs with ultrahigh capacity,but also provides new insights into engineering materials for energy storage applications.
Acknowledgements
The results described in this paper are obtained on the China National Grid (http://www.cngrid.org) and China Scientific Computing Grid(http://www.scgrid.cn).[50]
猜你喜欢
海外文摘·学术(2022年3期)2022-05-07
High Technology Letters(2021年4期)2022-01-09
作文大王·低年级(2020年12期)2020-12-31
娃娃乐园·综合智能(2020年12期)2020-12-26
娃娃乐园·综合智能(2020年11期)2020-11-14
娃娃乐园·综合智能(2020年5期)2020-06-07
37°女人(2019年9期)2019-12-16
中华诗词(2019年9期)2019-05-21
试题与研究·中考化学(2016年1期)2016-09-30
美与时代·城市版(2016年9期)2016-05-14

- Chinese Physics B的其它文章
- Speeding up generation of photon Fock state in a superconducting circuit via counterdiabatic driving∗
- Micro-scale photon source in a hybrid cQED system∗
- Quantum plasmon enhanced nonlinear wave mixing in graphene nanoflakes∗
- Restricted Boltzmann machine: Recent advances and mean-field theory*
- Nodal superconducting gap in LiFeP revealed by NMR:Contrast with LiFeAs*
- Origin of itinerant ferromagnetism in two-dimensional Fe3GeTe2∗