
芳香族L-氨基酸脱羧酶缺乏症一家系临床表现及遗传学分析
2021-05-01 13:57蔡慧强胡恕香蔡淑英彭桂兰
临床儿科杂志 2021年4期
蔡慧强 胡恕香 蔡淑英 彭桂兰
厦门大学附属妇女儿童医院 厦门市妇幼保健院儿童神经康复科(福建厦门 361003)
芳香族L-氨基酸脱羧酶缺乏症(aromatic L-amino acid decarboxylase deficiency,AADCD,OMIM:608643)是一种罕见的神经代谢疾病,为常染色体隐性遗传,因芳香族L-氨基酸脱羧酶(aromatic L-amino acid decarboxylase,AADC)活性降低,影响多巴胺、血清素等多种神经递质的合成,临床主要表现为动眼危象、肌张力障碍、发育迟缓和自主神经症状等[1]。AADCD 常在婴儿期发病,尚未明确基因型与临床表型相关性,临床诊断困难,常被误诊或漏诊。现回顾分析AADCD-家系的临床特征及基因检测结果,并进行文献复习。
1 临床资料
先证者,男,3 岁6 个月,系G 1 P 1,足月顺产,出生体质量3 450 g,出生时无窒息抢救史;生后易哭闹,难喂养。患儿3月龄时竖头不稳,肢体主动活动减少,肌张力低下,对“咯咯声”有反应,夜间睡眠易哭闹,多汗。患儿5月龄时查头颅磁共振成像(MRI):脑实质平扫未见异常,脑外间隙较增宽。视、听觉脑干诱发电位无异常。Gesell评估量表测试:大运动33,精细动作55,语言58,适应行为72,个人-社交52,总发育商54。患儿5 个半月时出现眼球运动异常,表现为突然双眼强直性外斜或偏斜,无口唇发绀、无肢体抽动,持续3~4小时,入睡后可缓解,间隔2~5天发作1次。发作期监测脑电图未见痫性放电。查血生化、血尿串联质谱(福州金域医学检验所检测)及染色体核型分析均未见异常。患儿11月龄时外院就诊,考虑阵发性非运动源性运动障碍,予口服氯硝西泮片,每次0.25 mg,每天2次,症状无改善。此次入院患儿眼球异常运动发作间隔2~5 天不等,每次发作最长可持续5~7小时;发育明显落后,不能维持竖头、坐位,无有意义语言;多汗,体质量无明显增长,夜间睡眠欠佳。入院体格检查:身长96 cm(P15),体质量12.5 kg(P3~P15),头围48 cm(P3~P15),神志清,倦怠面容,双肺呼吸音粗,心律齐,各瓣膜区未闻及杂音,腹软,肝脾肋下未触及,肌张力低下,膝腱反射弱,病理征阴性。父母均体健,非近亲结婚,否认类似家族遗传史。
先证者弟弟,男,3月龄,G2P2,足月顺产,出生体质量2 900 g,出生时无窒息缺氧抢救史;生后喂养、体质量增长尚可。患儿3月龄时可抬头,竖头稍不稳,可追视、追听,可逗笑。体格检查:神志清楚,头围40 cm(P15~P50),身长62 cm(P50~P85),体质量6.9 kg(P50~P85),前囟平软,肌张力稍低下,膝腱反射可引出,踝阵挛未引出,病理征阴性。
因为病因不明,经医院医学伦理审核,并征得父母知情同意后,抽取先证者及其父母外周血3 mL,送福州金域医学检验所作全外显子基因检测。采用QIAamp DNA提取试剂盒(QIAGEN公司)抽提基因组DNA,后进行PCR 扩增、杂交、捕获及纯化等,在Novaseq 6000 测序仪(Illumina Inc,USA)对基因组外显子区域进行测序。对可疑候选变异的位点设计PCR 引物进行扩增及Sanger 测序验证,明确变异来源。最终发现先证者携带DDC 基因c.714+4(IVS 6)A>T、c.1234(exon 13)C>T 复合杂合变异,前者来源于父亲,后者来源于母亲,符合常染色体隐性遗传。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)序列变异解读标准和指南[2]进行致病性判断,c.714+4(IVS 6)A>T 为内含子区的点变异,预计会引起剪接位点发生改变,编码的蛋白质发生紊乱而丧失其正常功能,多个在线软件预测其明显影响mRNA剪接,在千人基因组、dbSNP147数据库有收录(0.000199681,rs 200362242)。c.1234(exon 13)C>T 为错义变异,影响DDC 蛋白C 末端构象的正确折叠,在千人基因组、dbSNP 147 数据库有收录(0.000199681,rs542063660)。二者在ESP6500siv2_ALL数据库均未见收录。综合患儿临床表型,根据ACMG 致病性评级为可能致病性变异。先证者弟弟行一代测序,发现其携带与先证者同样的DDC 基因复合杂合变异c.714+4(IVS6)A>T、c.1234(exon13)C>T(图1和图2)。
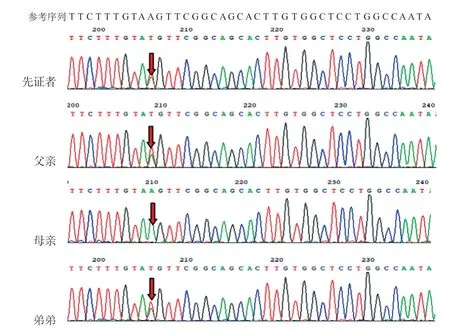
图1 DDC 基因c.714+4(IVS6)A>T 变异测序图
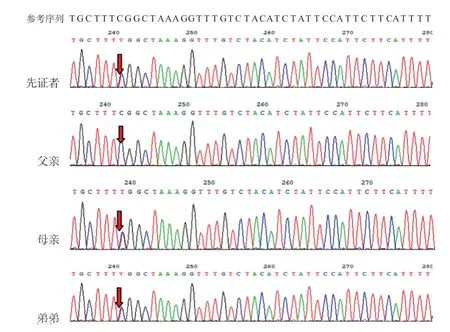
图2 DDC 基因c.1234(exon13)C>T 变异测序图
因我国大陆地区未能行血浆AADC 酶活性等检测,将先证者的血干滤纸片转送中国台湾省国立台湾大学医学院附属医院,经液相色谱-串联质谱法检测3-O-甲基多巴(3-OMD),结果为659.7 ng/mL(参考值<300 ng/mL),明显升高。随访过程中,先证者于4岁5个月因肺部感染死亡。先证者弟弟,5月龄出现夜间睡眠易哭闹,汗多,肢体自主活动减少,肌张力低下,竖头不稳、不会翻身;头颅MRI 示胼胝体体部稍纤细;6 月龄出现双眼强直性外斜或偏斜,无口唇发绀、无肢体抽动,无发热,每次持续1~4小时不等,间隔3~7天发作1次,发作期监测脑电图未见痫性放电;予口服维生素B6,50 mg/次,bid;普拉索克,0.01 mg/(kg·d),间隔7天增加0.005 mg/(kg·d),至0.03 mg/(kg·d)维持;司来吉兰,0.1 mg/(kg·d),间隔2 周增加至0.2 mg/(kg·d)维持;叶酸,5 mg/次,qd。现患儿仍有动眼危象发作,次数无减少,发育明显落后,不能竖头,肌张力低下,多汗,体质量不增。
2 讨论
AADC 又称为多巴脱羧酶(dopa decarboxylase,DDC),是一种合成神经递质5-羟色胺和多巴胺的终末酶。AADCD 是因编码DDC 基因变异,AADC 酶活力下降,左旋多巴和L-5-羟色氨酸脱羧障碍,继发多巴胺等神经递质合成不足,导致严重的神经功能障碍[1]。
DDC基因位于7p12.2染色体上,包含15个外显子,全长107 kb以上,内含子长度为1.0~17.0 kb[3],目前已报道DDC基因变异有50多种[4]。研究发现c.714+4(IVS 6)A>T 是中国台湾AADCD 最常见的基因型,占所有DDC变异的76%[5]。Dai等[6]报道的17例中国大陆AADCD患者中,3例c.714+4(IVS6)A>T纯合子变异,7例c.714+4(IVS6)A>T复合杂合变异,3例c.1234(exon13)C>T复合杂合变异。c.714+4(IVS6)A>T 为内含子剪接位点变异,可能导致DDC 基因剪接位点改变,编码蛋白功能异常。c.1234(exon 13)C>T 为错义变异,可影响DDC 蛋白C 末端构象的正确折叠。本例先证者及弟弟均携带DDC基因c.714+4(IVS 6)A>T、c.1234(exon 13)C>T 复合杂合变异,前者来源于父亲,后者来源于母亲,符合常染色体隐性遗传。
以“芳香族L-氨基酸脱羧酶缺乏症”、“AADCD”和“DDC”为检索词,分别检索PubMed、中国知网、万方数据库、人类基因组变异数据库,检索至2020 年6月,发现9例DDC基因c.714+4(IVS6)A>T、c.1234(exon13)C>T复合杂合变异患者[5-8]。临床特点:发病年龄3~4 月龄,临床表型严重,脑电图无特异性。AADCD 发病年龄常在1 岁内,平均诊断年龄为3.5岁[1]。本组2例患儿发病年龄在3月龄,发病时间相对较早。AADCD临床表现多样,最常见的症状是运动障碍(动眼危象、肌张力障碍、运动倒退)、全面发育迟缓、肌张力低下和自主神经症状(多汗、上睑下垂、鼻塞);不常见的表现有癫痫发作、易怒、行为异常、睡眠障碍、进食困难、间歇性低体温等[9]。根据运动障碍及发育迟缓严重程度,AADCD大致分为轻度,轻度发育里程碑延迟、轻度智力残疾;重度,无发育里程碑或非常有限的发育里程碑;中度介于两者之间[1]。本组2 例患儿临床表型均为重度,最终未能达到任何发育里程碑。临床上AADCD的动眼危象易与癫痫混淆,发生机制可能与脑内左旋多巴及其代谢物增加、去甲肾上腺素不足以及多巴胺能和胆碱能神经递质传递不平衡密切相关[10],建议行脑电图来鉴别,有利于及时抗癫痫治疗。
2017年AADCD诊治指南中指出诊断的3个核心要点,基因检测可作为确诊依据[1]。脑脊液神经递质和AADC酶活性检测是重要诊断依据,但临床中检测能力有限。Chien 等[11]对127 987 例新生儿行AADC缺陷筛查,其中4例新生儿的3-OMD浓度升高,后经基因检测均确诊为AADCD。另有研究通过电喷雾串联质谱(ESI-MS/MS)方法来量化3-OMD浓度,运用高通量检测进一步验证检测血滤纸3-OMD 浓度来识别AADCD 的可行性,表明血滤纸3-OMD 可作为AADCD辅助诊断之一[12]。本例先证者血滤纸3-OMD明显升高,基因检测携带了DDC基因c.714+4(IVS6)A>T、c.1234(exon13)C>T复合杂合变异,其弟弟有相似临床表现,且携带相同基因,诊断明确。
目前AADCD尚无特效治疗方法。指南建议使用选择性多巴胺受体激动剂、单胺氧化酶抑制剂和吡哆醇作为一线治疗用药,同时建议多药联合治疗[1]。理论上AADCD因左旋多巴过度甲基化为3-OMD而耗尽甲基供体,包括S-腺苷甲硫氨酸和四氢叶酸甲酯,可能继发大脑叶酸缺乏症,因此指南中指出AADCD可补充叶酸。AADCD平均病死年龄为4.6岁,多因肺炎、脓毒症、心力衰竭而死亡。本例先证者4岁5个月因肺部感染死亡。先证者弟弟给予多巴胺激动剂、维生素B6、单胺氧化酶抑制剂、叶酸联合治疗后,动眼危象无改善,未达到任何发育里程碑。近年来基因治疗给AADCD带来新的希望,通过腺病毒相关载体介导的DDC 基因进入壳核治疗,可恢复壳核质中的多巴胺水平,改善患者的运动、语言、认知功能[13-15]。也有研究显示基因治疗需在神经损伤发作前才有疗效,且对中度患者改善较明显[4]。但基因治疗目前存在很多不良反应[16],尚处于临床试验阶段,进展缓慢,是否能成为治疗AADCD 的有效方法有待进一步临床研究。
综上,AADCD 是一种罕见的神经代谢性疾病,临床表型非特异性,DDC基因c.714+4(IVS6)A>T、c.1234(exon13)C>T复合杂合变异的临床发病早,表型常为重度,预后差。对于疑似AADCD 或有家族病史,孕期可对绒毛或羊水细胞进行遗传分析,有利于产前遗传咨询,减少发病率。
猜你喜欢
中国食物与营养(2022年5期)2022-06-17
中国食物与营养(2022年5期)2022-06-17
中国康复(2021年6期)2021-11-30
家庭医学·下半月(2020年2期)2020-04-26
科学生活(2019年7期)2020-01-01
考试周刊(2017年26期)2017-12-12
家庭百事通·健康一点通(2017年8期)2017-08-18
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中国实用医药(2016年9期)2016-05-17
