
AMN 基因复合杂合变异致Imerslund-Gräsbeck 综合征1 例临床及基因分析
2021-05-01 11:21厉广栩孙薇薇陈秀灵
临床儿科杂志 2021年4期
厉广栩 潘 翔 逯 军 孙薇薇 陈秀灵
中南大学湘雅医学院附属海口医院儿科(海南海口 570208)
Imerslund-Gräsbeck综合征(Imerslund-Gräsbeck syndrome,IGS)是罕见的常染色体隐性遗传病,首次于1960年报道。IGS发病率低于6/1000000,临床特点是由维生素B12选择性吸收障碍造成巨幼细胞性贫血,部分可出现蛋白尿、反复感染及神经系统症状等。已知导致IGS 的主要原因是AMN 基因和CUBN 基因变异,以挪威、芬兰和地中海沿岸国家报道较多[1]。甲基丙二酸血症又名甲基丙二酸尿症,按发病原因可分为原发性甲基丙二酸血症和继发性甲基丙二酸血症。维生素B12缺乏是导致继发性甲基丙二酸血症发生的主要原因,补充维生素B12后,短期内临床症状可明显改善[2]。本文回顾分析1例由AMN基因复合杂合变异所致IGS继发甲基丙二酸血症患儿的临床特点和基因检测结果。
1 临床资料
男性患儿,10岁,因面色、口唇苍白,偶伴有呕吐就诊于中南大学湘雅医学院附属海口医院儿科。患儿系G3P3,足月顺产,出生体质量3.5 kg,无产伤及窒息史。生长发育与正常同龄儿相仿。患儿曾在3 岁时出现头发枯黄、双手指末梢发红。由于症状较轻,家长未予重视。父母均体健,非近亲结婚。患儿大姐有精神分裂症病史。体格检查:身高138.0 cm(P15~P50),体质量31 kg(P50),精神反应可;面色、口唇苍白,舌苔消失,头发枯黄,皮肤、黏膜无黄染和出血点,全身浅表淋巴结无肿大;头颅五官无畸形;心、肺、腹及神经系统无特殊;脊柱、四肢无畸形,双手指近甲根处皮肤呈红斑状改变。实验室检查:血红蛋白 84 g/L,红细胞2.44×1012/L,红细胞平均体积96.30 fL,网织红细胞百分比2.08%,白细胞1.87×109/L,中性粒细胞计数0.82×109/L;血维生素B12浓度83 pg/mL;血同型半胱氨酸77.71 μmmol/L,血电解质、肝肾功能、心肌酶谱无异常;尿蛋白无异常;血串联质谱示丙酰肉碱2.05 μM(0.50~4.00 μM),乙酰肉碱3.51 μM(6.00~30.00 μM),丙酰肉碱与乙酰肉碱比值增高;尿有机酸分析示甲基丙二酸20.3(0.0~4.0)。初步诊断为维生素B12缺乏症、巨幼细胞性贫血、中性粒细胞减少症、甲基丙二酸血症及同型半胱氨酸血症。
为进一步明确发病原因,经患儿家长知情同意和医院医学伦理委员会审查批准,抽取患儿及其父母外周静脉血各3 mL,送北京康旭医学检验所行遗传代谢基因包(panel)检测,排除原发性甲基丙二酸血症相关基因ACSF 3、MMUT、MMAA(cblA 型)、MMAB(cblB 型)、MMACHC(cblC 型)、MMADHC(cblD型)、LMBRDI(cdlF型)、MCEE、CD320、ALDH6A1、SUCLA2、SUCLG1、HCFC1、ABCD4存在致病性变异。进一步行全外显子测序及Sanger测序验证。结果显示,患儿第14号染色体上的AMN基因存在 c.527_530del杂合变异和 c.651+1G>C杂合变异,构成复合杂合变异。其中,c.527_530 del(编码区第 527_530 号核苷酸缺失)为移码变异,导致从第176 号氨基酸Asp 开始的氨基酸合成发生改变,并在改变后的第22 个氨基酸终止(p.Asp176GlyfsTer22);c.651+1G>C(编码区第 651号核苷酸后内含子中第1位核苷酸由G变为C)为剪切变异。HGMDPro、PubMed、千人基因组及dbSNP数据库等均未收录上述变异位点。上述变异经MutationTaster、Polyphen、SIFT等生物信息学软件预测均为有害变异;根据ACMG指南致病等级评估,AMNc.527_530del为疑似致病性变异(PVS1,PM2),AMNc.651+1 G>C 为致病性变异(PVS 1,PM 2,PS 2)。Sanger 测序验证发现患儿AMNc.527_530 del来源于其母亲,而AMNc.651+1 G>C 为新发变异(图 1)。结合患儿临床特点及基因变异检测结果,IGS诊断成立。
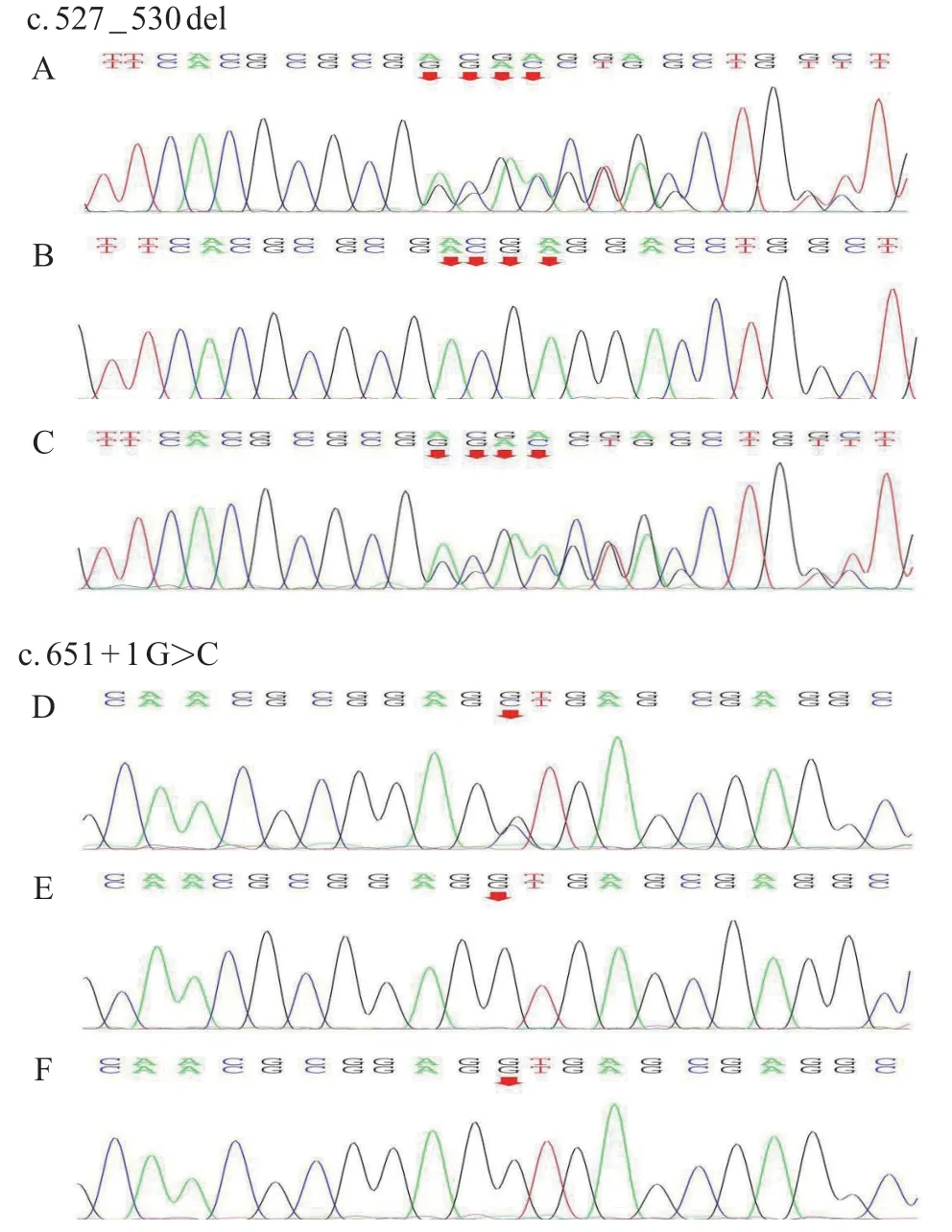
图1 患儿及父母AMN 基因Sanger 测序图
诊断明确后,单次给予1 mg 维生素B12肌注,患儿红细胞和白细胞计数逐渐回升。后期根据血常规及维生素B12浓度变化趋势,给予维生素B12肌注治疗,并嘱其加强营养摄入。目前患儿一般情况可,面色苍白及双手手指末梢发红症状缓解。复查血常规、尿常规、维生素B12浓度大致无异常,血串联质谱显示丙酰肉碱与乙酰肉碱比值正常,尿有机酸分析提示尿甲基丙二酸在正常范围内。
2 讨论
IGS 亦被称为维生素B12选择性吸收不良伴蛋白尿,是一种罕见的常染色体隐性遗传性疾病,以内因子治疗效果欠佳的巨幼细胞性贫血为主要临床特征,而对肠外维生素B12治疗有反应。大约50%的IGS患者会出现选择性低分子量蛋白尿,但未伴随其他肾病表现[3]。二代测序技术在临床推广前,IGS 和内因子缺乏所致巨幼细胞性贫血在临床上较难区分,部分IGS很可能被误诊。IGS相关变异基因有14号染色体的AMN和10号染色体的CUBN,分别编码amnionless亚基和cubilin 亚基,这两种亚基均为内因子-维生素B12复合体的重要组成部分,该复合体在回肠和肾近端小管均有分布[4]。回肠的内因子-维生素B12复合体负责维生素B12的吸收,而在肾近端小管的则负责蛋白质分子的重吸收。截止至2015年,人类基因变异数据库(The Human Gene Mutation Database,HGMD)收录的CUBN 基因致病性变异有37 个,AMN 基因致病性变异有30个[1]。
维生素B12参与红细胞生成、核酸合成、核糖体合成以及神经髓鞘形成等生理代谢。已知体内维生素B12可作为甲硫氨酸合成酶和L-甲基丙二酰辅酶A 变位酶的辅酶,前者可催化同型半胱氨酸甲基化为甲硫氨酸,与四氢叶酸再生和核酸合成有关;后者可催化甲基丙二酰辅酶A生成琥珀酰辅酶A,参与三羧酸循环。维生素B12缺乏可影响甲硫氨酸合成酶的功能,同型半胱氨酸向甲硫氨酸转化发生障碍,血同型半胱氨酸水平升高;另一方面导致核酸合成障碍,细胞分裂无法顺利进行,从而导致巨幼细胞性贫血。L-甲基丙二酰辅酶A 变位酶缺陷阻碍甲基丙二酰辅酶A转化为琥珀酰辅酶A,最终甲基丙二酰辅酶A 将通过旁路途径代谢为甲基丙二酸,而导致甲基丙二酸血症。
根据国外文献报道,IGS 的平均发病年龄为3.5岁,临床表现包括疲劳、面色苍白、生长发育迟缓、中性粒细胞减少、反复感染、神经系统病变、胃肠道功能紊乱、黄疸、心脏杂音和良性非特异性蛋白尿等,这些临床表现在维生素B12治疗后短期内很难消失[5]。同型半胱氨酸血症和甲基丙二酸血症间接反映了体内维生素B12处于缺乏状态。IGS 的发病与维生素B12的选择性吸收不良有关,一旦确诊,首选维生素B12肌肉注射治疗,每月1 次,具体用药方案还应参考年龄、临床症状严重程度和血维生素B12水平等因素。维生素B12维持治疗的IGS 患者远期预后一般较好。虽然相当数量的文献报道伴有持续蛋白尿的IGS患者肾功能长期维持正常,但亦有个别案例出现肾功能恶化[6],故应监测该类患儿尿蛋白和肾功能变化。
本例IGS 患儿因面色、口唇苍白就诊,经实验室检测考虑为维生素B12缺乏、巨幼细胞性贫血、中性粒细胞减少,还存在甲基丙二酸血症和同型半胱氨酸血症,考虑与维生素B12缺乏有关。遗传代谢基因包(panel)筛查排除原发性甲基丙二酸血症相关基因变异。完善全外显子测序后发现患儿第14 号染色体AMN 基因存在未见报道的致病性复合杂合变异(c.527_530 del 和c.651+1 G>C)。患儿明确诊断为IGS,经维生素B12肌注治疗后贫血症状明显好转,中性粒细胞计数恢复正常,精神反应佳,甲基丙二酸血症和同型半胱氨酸血症消失。随诊至今,患儿未出现蛋白尿症状,一般状态良好。
综上,IGS 是导致儿童巨幼细胞性贫血的原因之一,且并非所有的IGS 患儿均有蛋白尿的临床表现,及时完善基因检测有利于明确发病原因,使治疗更为精准化,亦有利于改善患儿预后。
猜你喜欢
分子催化(2022年1期)2022-11-02
石油化工(2022年9期)2022-10-19
中老年保健(2022年4期)2022-08-22
昆明医科大学学报(2021年1期)2021-02-07
祝您健康(2020年5期)2020-05-14
健康博览(2020年2期)2020-02-27
华声文萃(2019年4期)2019-09-10
文萃报·周二版(2019年10期)2019-09-10
中国医药导报(2011年27期)2011-12-31
中学理科·综合版(2008年11期)2008-01-14
