
曲咪新乳膏的有关物质研究
2021-04-30 02:05:46古丽霞廖志刚邓红林华庆梁勇坤余良钟张蜀赖昌威
广东药科大学学报 2021年2期
古丽霞,廖志刚,邓红,2,林华庆,梁勇坤,余良钟,张蜀,2,赖昌威
(1.广东药科大学广东省药物新剂型重点实验室,广东广州510006;2.广东药科大学广东省局部精准药物递药制剂工程技术研究中心,广东广州510006;3.广东华润顺峰药业有限公司,广东佛山528300)
曲咪新乳膏为外用复方制剂,曾用名为皮康霜,每克乳膏中含有1 mg醋酸曲安奈德、10 mg硝酸咪康唑、3 000 U 硫酸新霉素;临床上主要用于过敏性皮炎、湿疹、失禁相关性皮炎、体癣、股癣以及手足癣等病症治疗[1-4]。
曲咪新乳膏现行标准有《国家药品标准》化学药品地标升国标第二册、1996 年版北京市药品标准及企业内控标准,现有标准未对有关物质作出要求,主要针对醋酸曲安奈德鉴别和含量测定作了规定:以氯化三苯四氮唑为显色剂,紫外-可见光分光光度计测定其含量[5-6]。该方法稳定性差,前处理繁琐,检测结果受环境影响,且无法测定主药的有关物质[7-8]。
目前,曲咪新乳膏质量研究主要集中在3 种主药的含量测定,已有文献报道采用固相萃取-HPLC法、管碟法、HPLC-ELSE 法同时测定1 种或者2 种主药含量[9-11]。梁秋霞等[12]简述了曲咪新乳膏3种主药的有关物质和含量测定方法,其侧重点在于对大量企业抽检样品进行测定,评估市场上曲咪新乳膏总体质量。本研究系统筛选了有关物质供试品溶液的制备方法,通过强制降解试验优化了色谱条件,并探讨曲咪新乳膏主药的降解途径,所建立的UHPLC-PDA 法和溶剂萃取-高速离心-HPLC-ELSE法能有效检测出乳膏的有关物质,为本品质量控制提供参考。
1 仪器与试药
AcQuity 超高效液相色谱仪(配制PDA 检测器,美国Waters 公司);2695 型高效液相色谱仪(美国Waters 公司);2420 型ELSD 检测器(美国Waters 公司);色谱柱为Thermo scientific AccucoreaQ C18(100 mm×2.1 mm,2.6 μm),Kromasil C18(250 mm×4.6 mm,5 μm);TGL-20B高速离心机(上海安亭科学仪器厂);BSA224S-CW 分析天平、CP225D 分析天平(德国赛多利斯公司)。
曲咪新乳膏(广东华润顺峰药业有限公司,批号:20151111、20151112、20151113、20160601、20160602、20160603、20161201、20161202、20161203、20170301、20170302、20170303、20170601、20170602、20170603、20170808、20170810、20170812、20171201、20171202、20171203;A 企业,批号:160203、170403;B 企业,批号:B1058;C 企业,批号:170304;D 企业,批号:20170908;E 企业,批号:170904);原料药:醋酸曲安奈德(天津天药药业股份有限公司,批号:NTAC170701;质量分数98.5%),硝酸咪康唑(广州市汉普医药有限公司,批号:20170301;质量分数:100.5%),硫酸新霉素(宜昌三峡制药有限公司,批号:201708034,每1 mg 的效价为667 个单位,水分:4.5%);对照品:曲安奈德(批号:100055-201103,质量分数:98.8%),新霉胺(批号:130411-200908)均购自中国食品药品检定研究院;甲醇、乙腈、四氢呋喃为色谱纯;其他试剂为分析纯;水为屈臣氏蒸馏水。
2 方法与结果
2.1 曲安奈德及其他有关物质
2.1.1 溶液配制 供试品溶液:取本品约2.5 g(约相当于醋酸曲安奈德2.5 mg、硝酸咪康25 mg),精密称定,置10 mL 量瓶中,加入甲醇-四氢呋喃(体积比1∶1,下同)适量,在80 ℃水浴分散2 min,放冷至室温,加甲醇-四氢呋喃稀释至刻度,轻摇匀,冰浴2 h,取出迅速经微孔滤膜过滤,取续滤液,即得。
对照品溶液:取曲安奈德对照品约12.5 mg,精密称定,置50 mL 量瓶中,加甲醇-四氢呋喃振摇溶解并稀释至刻度,摇匀,精密量取该溶液与供试品溶液各l mL,置同一100 mL 量瓶中,用甲醇-四氢呋喃稀释至刻度,摇匀,即得。
2.1.2 色谱条件与系统适用性 AcQuity 超高效液相色谱仪(配制PDA检测器,美国Waters公司);色谱柱为Thermo scientific AccucoreaQ C18(100 mm×2.1 mm,2.6 μm);流动相A 为甲醇-乙腈-1%乙酸铵(体积比23∶23∶54),流动相B 为甲醇-乙腈-1%乙酸铵(体积比40∶40∶20),采用梯度洗脱(0~8 min,100%A;8~16 min,100%A→0%A;16~26 min,0%A;26~28 min,0%A→100%A);流速:0.5 mL/min;检测波长:235 nm;柱温:30 ℃;进样量:10 μL。取“2.1.1”项供试品溶液与对照品溶液依法进样,记录色谱图见图1,可见曲安奈德峰与醋酸曲安奈德峰的分离度应大于10.0。
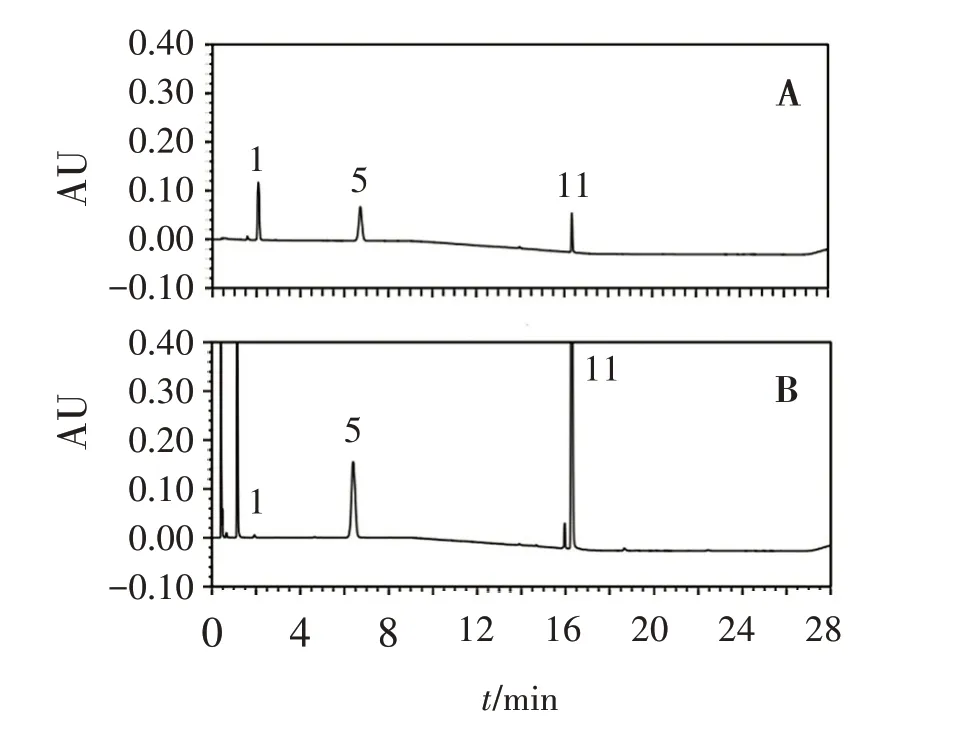
图1 醋酸曲安奈德、硝酸咪康唑对照品溶液(A)与供试品溶液(B)色谱图Figure 1 Chromatograms of triamcinolone acetonide acetate,miconazole nitratesystem control solution (A) and test solution(B)
2.1.3 强制降解实验 取曲咪新乳膏(顺峰,批号:20170808;A 企业,批号:160203)、缺醋酸曲安奈德阴性乳膏(自制)、缺硝酸咪康唑阴性乳膏(自制)、全空白乳膏各2.5 g,醋酸曲安奈德原料药2.5 mg,硝酸咪康唑原料药25 mg分别置10 mL量瓶;按下列方法处理后,加甲醇-四氢呋喃适量,80 ℃水浴分散2 min,放冷,加混合溶剂稀释至刻度,冰浴2 h,摇匀,滤过,即得。
(1)酸强制降解试验:加6 mol/L HCl溶液1 mL,放置10 h,加6 mol/L NaOH溶液1 mL。
(2)碱强制降解试验:加6 mol/L NaOH 溶液1 mL,放置30 min,加6 mol/L HCl溶液1 mL。
(3)氧化强制降解试验:加30%H2O2溶液1 mL,放置72 h。
(4)高温强制降解试验:烘箱110 ℃放置24 h,放至室温。
(5)光照强制降解试验:采用光照试验仪(4 500 lx)照144 h。
分别取上述未破坏和各强制降解后溶液,按“2.1.2”项色谱条件进样,记录色谱图,结果见图2。
可见,曲咪新乳膏降解前可检测出有关物质曲安奈德;各降解条件下,较明显的降解产物均为曲安奈德。乳膏在碱性条件下较不稳定,除曲安奈德外,同时产生1 个醋酸曲安奈德降解产物(峰2)和1个硝酸咪康唑降解产物(峰4);氧化降解阴性乳膏(自制)和供试品乳膏均检测出峰13,推测为辅料降解产物。各主药与降解产物峰归属见表1。

图2 醋酸曲安奈德和硝酸咪康唑样品溶液强制降解试验色谱图Figure 2 Forced degradation test chromatograms of sample solutions of triamcinolone acetonide acetate and miconazole nitrate
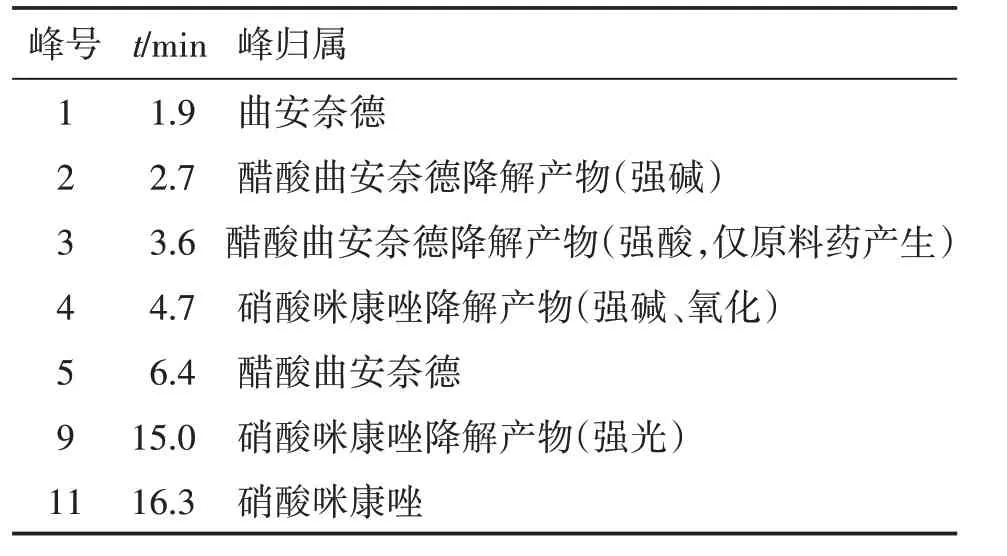
表1 主药与降解产物峰归属Table 1 Peak assignment of main drug and degradation products
2.1.4 溶液稳定性 取“2.1.1”项供试品溶液和对照品溶液,于室温自然光下放置0、1、2、4、6、8 h 后取出,进样测定,供试品溶液可检出曲安奈德,峰面积RSD 为2.16%,未见新杂质产生;对照品溶液曲安奈德、醋酸曲安奈德、硝酸咪康唑峰面积RSD 为2.34%、2.87%、3.39%,表明供试品溶液和对照品溶液在8 h内稳定。
2.1.5 检测限 取“2.1.1”项对照品溶液,逐级稀释,进样测定,按信噪比为3∶1计算检测限,曲安奈德的检测限为0.24 μg/mL、醋酸曲安奈德为0.42 μg/mL、硝酸咪康唑为3.54 μg/mL。
2.1.6 有关物质样品测定 取本品,依法测定,记录色谱图,结果曲安奈德为最大单个杂质,按外标法以峰面积计算均不超过1.0%;样品中归属硝酸咪康唑的相关杂质,均未超过对照溶液主峰面积0.5%;总杂质均不超过对照溶液主峰面积1.5%。
2.2 硫酸新霉素有关物质新霉胺
2.2.1 溶液配制 供试品溶液:取本品适量(约相当于硫酸新霉素7.5 mg),精密称定,置25 mL 离心管中,加入三氯甲烷0.5 mL,强力振摇使溶解,离心30 min(10 000 r/min),分取水层置于5 mL量瓶中;再加水约1 mL 于离心管,振摇萃取,离心30 min(10 000 r/min),分取水层,重复以上操作3 次,合并水层于5 mL 量瓶,用水稀释至刻度,即得供试品溶液(每1 mL溶液含新霉素1.5 mg)。
新霉胺对照品溶液:取新霉胺对照品约15 mg,精密称定,置50 mL 量瓶中,加水稀释至刻度,作为新霉胺对照品储备液(0.3 mg/mL)。精密量取上述对照品储备液1 mL,置10 mL量瓶中,加水稀释至刻度,作为新霉胺对照品溶液(30 μg/mL)。
系统适用性溶液:取硫酸新霉素原料药适量(约相当于新霉素15 mg),精密称定,置10 mL 量瓶中,再精密加入上述对照品储备液1 mL,加水稀释至刻度,配制硫酸新霉素和新霉胺混合对照品溶液作为系统适用性溶液。
2.2.2 色谱条件与系统适用性 2695 型高效液相色谱仪(美国Waters 公司),2420 型ELSD 检测器(美国Waters 公司);色谱柱为Kromasil C18(250 mm×4.6 mm,5 μm);流动相为甲醇-含0.7%五氟丙酸的20 mmol/L 乙酸铵溶液(体积比55∶45);流速:1 mL/min;进样量:20 μL;柱温:30 ℃;雾化温度:60 ℃;漂移管温度:70 ℃;载气压力:206.85 kPa;撞击器:关;增益值:10。取“2.2.1”项混合对照品溶液,依法进样,记录色谱图见图3,可见新霉素峰保留时间约为8 min。
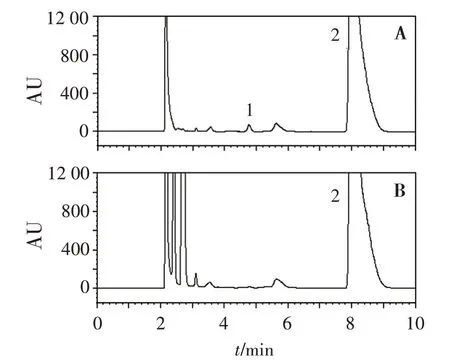
图3 新霉素系统适用性溶液(A)与供试品溶液(B)色谱图Figure 3 Chromatograms of neomycin system suitability solution(A)and sample solution(B)
2.2.3 强制降解试验 取曲咪新乳膏(顺峰,批号:20170812;A 企业,批号:170403)、缺硫酸新霉素阴性乳膏(自制)、全空白乳膏各2 g,硫酸新霉素原料药7.5 mg,分别置25 mL 离心管,加入0.5 mL 三氯甲烷,振摇使溶解,按下列方法处理。
(1)酸强制降解试验:加入2 mol/L HCl 溶液1 mL,超声振摇1 h,放置过夜,加入1 mL 2 mol/L NaOH 溶液,加水约1 mL,振摇,离心30 min(10 000 r/min),取水层于5 mL量瓶并稀释至刻度。
(2)碱强制降解试验:加入2 mol/L NaOH 溶液1 mL,超声振摇1 h,加入1 mL 2 mol/L HCl 溶液,加水约1 mL,振摇,离心30 min(10 000 r/min),取水层于5 mL量瓶并稀释至刻度。
(3)氧化强制降解试验:加入三氯甲烷0.5 mL,振摇使溶解,加入1 mL 30% H2O2溶液,超声振摇1 h,按“2.2.1”项供试品溶液自“加水约1 mL…”制备。
(4)高温强制降解试验:加入三氯甲烷0.5 mL,振摇使溶解,沸水浴1 h,放冷并补足失重,按“2.2.1”项供试品溶液自“加水约1 mL…”制备。
(5)光照强制降解试验:加入三氯甲烷0.5 mL,置光照试验仪(4 500 lx)于2 h,放冷并补足失重,按“2.2.1”项供试品溶液自“加水约1 mL…”制备。
分别取上述未破坏和各强制降解后溶液,按“2.2.2”项色谱条件进样,记录色谱图,结果见图4。可见,曲咪新乳膏降解前新霉胺均符合限量要求;各降解途径新霉胺均无明显增加。
2.2.4 加标回收率 精密称取曲咪新乳膏(批号:20170810)约2.5 g 于25 mL 离心管中,加入“2.2.1”项下的新霉胺对照溶液0.5 mL,自加入“三氯甲烷0.5 mL”起,按“2.2.1”项供试品溶液的制备方法制备加标样品(6份),计算加标样品的回收率为100.9%,RSD为1.99%。
2.2.5 溶液稳定性 由于供试品未检出新霉胺,故采用加标样品进行溶液稳定性试验:取“2.2.4”项下的一份加标样品,室温放置,于0、1、2、4、6、8 h 测定,记录新霉胺的峰面积,计算RSD 为3.43%,表明溶液在8 h内基本稳定。
2.2.6 检测限 精密称取新霉胺对照品适量,逐级稀释,进样测定,按信噪比为3∶1 计算检测限,新霉胺检测限为6 μg/mL,即在供试品溶液浓度为1.5 mg/mL,含0.4%的新霉胺即被检出,满足ChP2020 硫酸新霉素中新霉胺的限度为2.0%的要求。
2.2.7 有关物质样品测定 按“2.2.2”项下色谱条件,注入色谱仪,记录色谱图,代表性图谱见图3,结果均无检出新霉胺。
3 讨论
3.1 曲安奈德及其他有关物质
3.1.1 供试品溶液制备溶剂和方法的考察 醋酸曲安奈德在三氯甲烷中溶解,硝酸咪康唑略溶于甲醇。根据乳膏和主药的溶解性,参考文献[13-15],本研究对甲醇、异丙醇、二氯甲烷、三氯甲烷、四氢呋喃及其两两组合进行筛选,结果显示三氯甲烷-甲醇(体积比1∶9)和甲醇-四氢呋喃(体积比1∶1)可分散曲咪新乳膏且容易滤过,使用其他溶剂出现浑浊或者分层,或无法过滤;进一步比较2 种混合溶剂,结果显示甲醇-四氢呋喃(体积比1∶1)溶解快且完全,因此选择甲醇-四氢呋喃(体积比1∶1)作为乳膏中醋酸曲安奈德和硝酸咪康唑的有关物质测定供试品溶液制备的溶剂。参考2020 年版《中国药典》(ChP2020)中醋酸曲安奈德乳膏含量测定项下供试品溶液的制备方法“置80 ℃水浴中加热2 min”,考察了醋酸曲安奈德和硝酸咪康唑的回收率分别为99.87%、102.09%,且未产生明显降解产物,说明80 ℃水浴不影响有关物质测定。
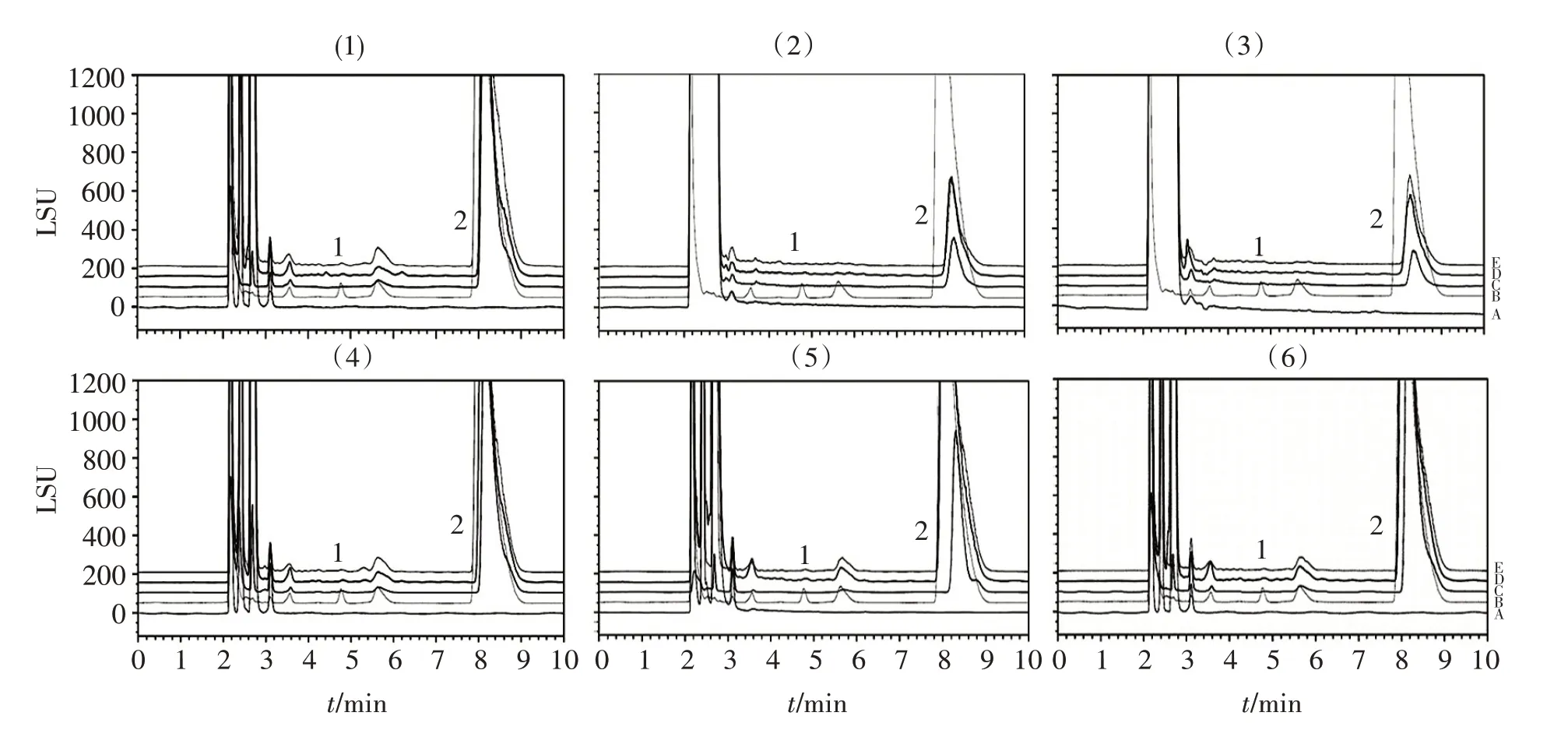
图4 硫酸新霉素样品溶液强制降解试验液相色谱图Figure 4 Forced degradation test chromatograms of neomycin sulfate sample solutions
3.1.2 色谱条件的选择 参考文献及各国药典,醋酸曲安奈德的流动相系统均采用甲醇-水或乙腈-水;硝酸咪康唑的流动相系统均采用甲醇-乙腈-乙酸铵[16]。经试验,甲醇-水或乙腈-水系统(不加乙酸铵)硝酸咪康唑无法出峰,因此,拟采用甲醇-乙腈-乙酸铵系统,同时测定乳膏中醋酸曲安奈德和硝酸咪康唑的有关物质。
ChP2020 中醋酸曲安奈德检测波长为240 nm,硝酸咪康唑为230 nm,BP2018咪康唑乳膏为235 nm,USP42~NF37 硝酸咪康唑乳膏为225 nm[17-19]。采用二极管阵列检测器进行扫描,发现波长为235 nm 时基线较平稳,可同时检测230 nm(硝酸咪康唑)和240 nm(醋酸曲安奈德)的有关物质,并可在两者之间获得相对平衡的响应值,因此选择检测波长为235 nm。
3.1.3 强制降解试验 本研究曾选择较温和的条件进行破坏,本品稳定,逐步加剧破坏条件至有明显的降解产物。强制降解试验显示,乳膏在碱性条件下较不稳定,加碱30 min 即需终止试验,并产生较多降解产物。高温强制降解试验曾尝试加入溶剂甲醇-四氢呋喃,沸水浴加热破坏,结果溶剂很快挥干,且乳膏制剂经处理后无法滤过;后采用将样品直接放入烘箱110 ℃、24 h,结果原料药稳定,制剂中曲安奈德峰增大,其中缺硝酸咪康唑阴性乳膏醋酸曲安奈德几乎完全降解为曲安奈德。原因可能是乳膏为半固体制剂,原料药为固体,固体状态下物质较为稳定。成品制剂比抽离硝酸咪康唑阴性乳膏稳定,说明处方工艺的合理性。
3.1.4 有关物质限度 ChP2020 中醋酸曲安奈德原料药曲安奈德的限量为1.0%,总杂质限度为2.5%;硝酸咪康唑原料药总杂质限度为0.5%。经强制降解试验和对多批样品的检测,本品主要降解产物为曲安奈德,暂定本品曲安奈德的限量为1.0%,其他最大单个杂质不得超过1.0%,总杂质不得超过3.0%。由于乳膏处方使用了较多辅料,且采用了梯度洗脱的色谱条件,有关物质的检查和计算需排除辅料峰和溶剂峰。
3.2 硫酸新霉素有关物质新霉胺
3.2.1 供试品溶液制备方法的考察 乳膏属半固体制剂,分油水两相,前期研究直接加水超声或加入温水后温水浴,体系呈乳浊状,无法过滤,因此先加入三氯甲烷强力振摇使油、水相分离,后以水洗涤油相使提取完全,硫酸新霉素在水中极易溶解,离心分离后合并水层为供试品溶液。
3.2.2 色谱条件的选择 各国药典中硫酸新霉素主要采用薄层色谱法检查新霉胺的限量。本研究曾参考ChP2020、BP2018[18]和李晓鑫[20]的薄层色谱条件进行检测,结果显示供试品溶液斑点过载明显,新霉胺斑点显色时容易化开,重现性差。参考戴向东[21]HPLC-ELSE 法,在此基础上优化色谱条件,发现乙酸铵降至20 mmol/L 时,新霉素的保留时间略增加;五氟丙酸降至0.7%时,新霉素的保留时间不变,优化后的色谱条件离子对试剂和缓冲盐用量更少。
3.2.3 强制降解条件的选择 新霉素在酸、碱和高温试验中均明显降解,但新霉胺仍未超过限度,说明新霉胺并非其主要降解产物,在原料药控制限度即可。新霉素主要成分为新霉素A(新霉胺)、新霉素B、新霉素C,其中新霉素C 效价约为新霉素B 的二分之一,因此EP9.0 和BP2018 中还规定了新霉素C 峰面积应为新霉素B 的3%~15%[22];查阅资料可知新霉素还含有巴龙霉胺、巴龙霉素Ⅰ和Ⅱ、新霉素的单-N-乙酰基衍生物(neomycin LP-A、LP-B 和LP-C)等有关物质[23-24],有待进一步研究。
4 总结
本研究收集了市售5个其他企业生产的曲咪新乳膏,从性状、离心稳定性及有关物质3 个方面比较,选择了质量最优的A 企业产品作为对照药品同时进行强制降解试验,建立了UHPLC-PDA 法和高速离心-HPLC-ELSE 法测定曲咪新乳膏的有关物质,结果表明方法专属性强,灵敏度高,可有效分离曲咪新乳膏3 种主药的有关物质,为本品质量控制提供参考。
猜你喜欢
中华耳科学杂志(2021年4期)2021-09-01 01:25:58
中国民间疗法(2021年1期)2021-04-20 02:31:12
中国抗生素杂志(2019年3期)2019-03-29 05:21:20
中国医疗美容(2015年4期)2015-04-27 02:24:13
西南军医(2015年3期)2015-04-23 07:28:28
中国医药科学(2015年2期)2015-02-27 12:32:12
中国实验诊断学(2014年10期)2014-06-01 12:30:52
西南军医(2014年5期)2014-04-25 07:42:36
河南医学研究(2014年3期)2014-02-27 14:51:55
中医研究(2013年5期)2013-03-11 20:26:56
