
Torsades de pointes episode in a woman with high-grade fever and inflammatory activation: A case report
2021-04-27 07:48HuiQiuHongWeiLiShuHongZhangXiaoGeZhouWeiPingLi
World Journal of Clinical Cases 2021年12期
Hui Qiu, Hong-Wei Li, Shu-Hong Zhang, Xiao-Ge Zhou, Wei-Ping Li
Hui Qiu, Hong-Wei Li, Wei-Ping Li, Department of Cardiology, Beijing Friendship Hospital,Capital Medical University, Beijing 100050, China
Shu-Hong Zhang, Xiao-Ge Zhou, Department of Pathology, Beijing Friendship Hospital, Capital Medical University, Beijing 100050, China
Abstract BACKGROUND QT interval prolongation can induce torsades de pointes (TdP), a potentially fatal ventricular arrhythmia. Recently, an increasing number of non-cardiac drugs have been found to cause QT prolongation and/or TdP onset. Moreover, recent findings have demonstrated the key roles of systemic inflammatory activation and fever in promoting long-QT syndrome (LQTS) and TdP development.CASE SUMMARY A 30-year-old woman was admitted with a moderate to high-grade episodic fever for two weeks. The patient was administered with multiple antibiotics after hospitalization but still had repeating fever and markedly elevated C-reactive protein. Once after a high fever, the patient suddenly lost consciousness, and electrocardiogram (ECG) showed transient TdP onset after frequent premature ventricular contraction. The patient recovered sinus rhythm and consciousness spontaneously, and post-TdP ECG revealed a prolonged QTc interval of 560 ms.The patient’s clinical manifestations and unresponsiveness to the antibiotics led to the final diagnosis of adult-onset Still’s disease (AOSD). There was no evidence of cardiac involvement. After the AOSD diagnosis, discontinuation of antibiotics and immediate initiation of intravenous dexamethasone administration resulted in the normal temperature and QTc interval. The genetic analysis identified that the patient and her father had heterozygous mutations in KCNH2 (c.1370C>T) and AKAP9 (c.7725A>C). During the 2-year follow-up period, the patient had no recurrence of any arrhythmia and maintained normal QTc interval.CONCLUSION This case study highlights the risk of systemic inflammatory activation and antibiotic-induced TdP/LQTS onset. Genetic analysis should be considered to identify individuals at high risk of developing TdP.
Key Words: Torsades de pointes; Long QT syndrome; Adult-onset Still’s disease;Antibiotics; Inflammation; Case report
INTRODUCTION
Torsades de pointes (TdP) is a rare but distinctive form of arrhythmia or polymorphic ventricular tachycardia characterized by the abnormal twisting of the QRS complex peaks around the isoelectric line. It has been well established that several pathological conditions and even drug toxicity can induce prolonged or abnormal repolarization(QT interval prolongation and/or abnormally twisted T or T/U wave morphology),resulting in TdP development, which in turn leads to ventricular fibrillation and sudden cardiac death. QT intervals represent the duration of the total action potential during the ventricular polarization and depolarization events. Essentially, long QT syndrome (LQTS) can be categorized as congenital (cLQTS) or acquired (aLQTS), and either of these can cause TdP pathogenesis. Recently, an increasing number of drugs,especially non-cardiac drugs that modulate cardiac repolarization, have been found to be associated with prolonged QT interval and occasional TdP onset[1]. It has been revealed that both aLQTS and cLQTS may share similar genetic variants for their pathogenesis[2-4]. In this context, the individual genetic background could be an important player, even in the presence of slight QT interval prolongation, because in association with appropriate pathogenic triggers, it may aggravate the life-threatening manifestations of aLQTS[5]. Here, we present the case study of TdP episode in a 30-year-old woman who underwent multiple courses of antibiotic treatments for highgrade fever of unexplained origin until diagnosed with adult-onset Still’s disease(AOSD), a rare systemic inflammatory disease with unknown etiology. The Yamaguchi criteria are the most popular of several proposed diagnostic criteria for AOSD, with fever of approximately 39 °C for at least a week, typical salmon colored rash, arthralgia or arthritis for at least 2 wk, and leukocytosis as major criteria, and sore throat, lymphadenopathy, liver dysfunction, negative rheumatoid factor, and antinuclear antibody as minor criteria[6].
CASE PRESENTATION
Chief complaints
A 30-year-old woman was admitted with a moderate to high-grade fever (maximum of 40.5 °C) lasting for two weeks.
History of present illness
The patient complained of sore throat and skin rashes in the first three days and had no shortness of breath, chest pain, cough, expectoration, hemoptysis, amaurosis, or arthralgia.
History of past illness
The patient had no considerable relevant medical history.
Personal and family history
The patient had no family history of either cardiovascular disease or sudden death.
Physical examination
On physical examination, the patient’s body temperature was recorded 38.2 °C, heart rate was 115 beats per min, respiratory rate was 23 breaths per minute, and blood pressure was 90/60 mmHg. The cardiopulmonary examination was unremarkable.
Laboratory examinations
Initial laboratory tests showed moderate leukocytosis (15.83 × 109/L) with predominant neutrophils (92.5%), anemia (hemoglobin 98 g/L), significantly elevated C-reactive protein (CRP, > 160 mg/L), erythrocyte sedimentation rate (95 mm/h), and serum ferritin (1362.7 ng/mL), and slightly elevated troponin I (0.116 ng/mL) along with normal creatine kinase-MB (CK-MB). Serum electrolyte examinations exhibited normal levels of potassium (3.74 mmol/L), sodium (133.1 mmol/L), and calcium (1.91 mmol/L). All relevant microbial cultures and specific antibody testing for infectious agents were also negative. An autoantibody panel including anti-nucleic acid, antidouble stranded deoxyribonucleic acid, anti-ENA, anti-neutrophil cytoplasmic, antirheumatoid factor, and anti-streptolysin O antibodies was tested negative. Cervical lymph node biopsy indicated symptoms of lymphocytic hyperplasia. The follow-up immunohistochemical analysis revealed that lymphocytic proliferation was mainly due to the T cell proliferation. Bone marrow puncture indicated that the proliferation of granulocytes was significantly active, and the proportions of intermediate and advanced granulocyte stages were significantly increased. Cellular morphology analysis suggested infection-associated reactive hyperplasia of the granulosa.
Imaging examinations
Abdominal ultrasonography exhibited the signs of splenomegaly. The 12-lead electrocardiogram (ECG) showed a regular sinus rhythm with a normal QTc of 440 ms.Transthoracic echocardiography demonstrated a normal cardiac structure and left ventricular ejection fraction of 71%. The chest X-ray examination suggested suspected pneumonia.
FINAL DIAGNOSIS
The patient was administered with multiple antibiotics including imipenem-cilastatin sodium, azithromycin, norvancomycin, moxifloxacin hydrochloride, and voriconazole within 2 wk, but still had repeating fever. During an episode of high fever and chills,the patient experienced palpitations. Immediate ECG examination indicated the presence of frequent premature ventricular contractions (PVCs) (Figure 1A). After 2 min, the patient suddenly lost consciousness, and ECG showed TdP onset at that point(Figure 1B). About 20 s later, she regained consciousness spontaneously, while post-TdP ECG revealed a sinus rhythm with a prolonged QTc interval of 560 ms (Figure 1C)and slightly reduced serum potassium level (3.50 mmol/L) compared to the previous serum test. The biochemical markers, including N-terminal pro-B-type natriuretic peptide, troponin I, CK-MB, aspartate aminotransferase, and alanine aminotransferase,were not elevated at this stage. However, CRP level remained extremely high (> 160 mg/L) when TdP occurred. Repeating episodes of PVCs were recorded immediately after infusion of linezolid to treat pneumonia, and that disappeared after initiating the treatment with intravenous lidocaine. Clinical manifestations of a spiking fever, skin rash, leukocytosis, elevated ferritin levels, splenomegaly, and unresponsiveness to multiple broad-spectrum antibiotics led to the final diagnosis of AOSD.
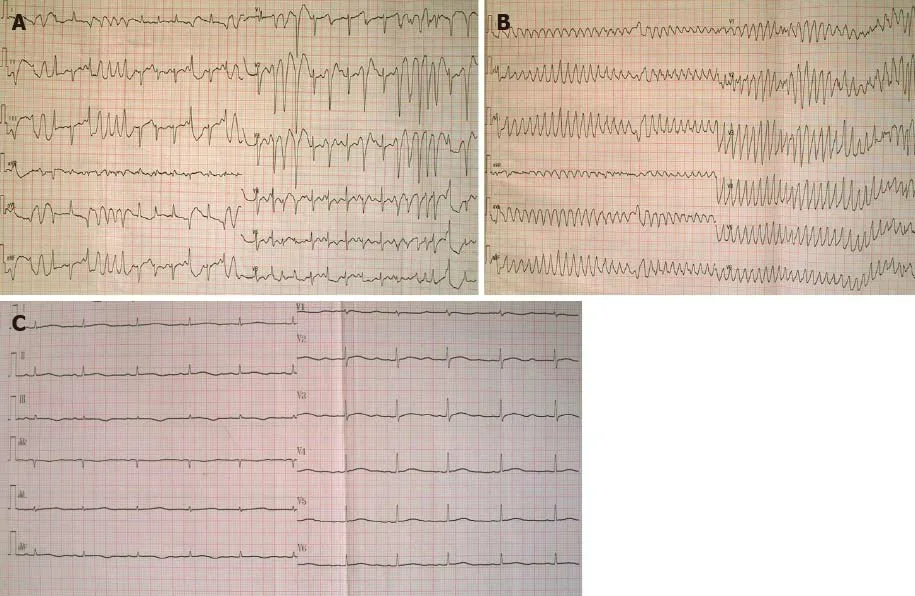
Figure 1 Electrocardiogram before and after Torsades de pointes. A: Electrocardiogram (ECG) exhibited frequent premature ventricular contractions(PVCs) during an attack of high fever and chills; B: ECG when the patient suddenly lost consciousness showed Torsades de pointes (TdP) after PVCs; C: ECG after TdP onset showed a sinus rhythm with a prolonged corrected QT interval of 560 ms.
TREATMENT
After the diagnosis of AOSD, all antibiotic therapies were stopped and intravenous dexamethasone (5 mg) administration was initiated once daily for two days, which helped quickly restore the normal body temperature and white blood cell count.Dexamethasone therapy exhibited rapid improvement of the elevated CRP level to the normal range (9 mg/L) in 5 d. The 24-h ambulatory ECG monitoring 2 d after the TdP episode exhibited a normal sinus rhythm and 25 ventricular premature beats.Moreover, the QTc interval was gradually shortened to the normal range (444 ms), and serious ventricular arrhythmia did not recur. Cardiac magnetic resonance imaging(CMRI) showed normal cardiac structure and function without early and late gadolinium enhancement. To further explore the possible mechanism underlying TdP onset, we performed the genetic testing with the patient’s and her immediate family member’s blood samples. Peripheral blood samples were collected from the patient,her mother, and her father. Total genomic DNA was extracted from peripheral blood samples of all subjects using a blood DNA extraction kit (TIANGEN, Beijing, China).Targeted gene capture sequencing was performed by MyGenostics (Beijing, China).Briefly, biotinylated capture probes were designed for the exons of 74 genes related to the cardiovascular system, and then sequencing was performed using Illumina HiSeq 2000 Next-Generation Sequencing platform, and the sequencing data were analyzed by a professional bioinformatics analyst (MyGenostics, Beijing, China). Clinically relevant genetic variants from the proband and parental samples were confirmed by Sanger sequencing. The genetic inheritance analysis further verified that the patient and her father, with a QTc interval of 490 ms, had heterozygous mutations in the genesKCNH2(c.1370C>T) andAKAP9(c.7725A>C), while her mother, with a normal QTc interval, had no relevant genetic mutations detected (Figures 2 and 3).
OUTCOME AND FOLLOW-UP
The patient was discharged 23 d after the admission without any further instructions to continue corticosteroid therapy. During the 2-year follow-up, the patient did not experience any sort of recurrence of arrhythmia and consistently maintained a normal QTc interval.
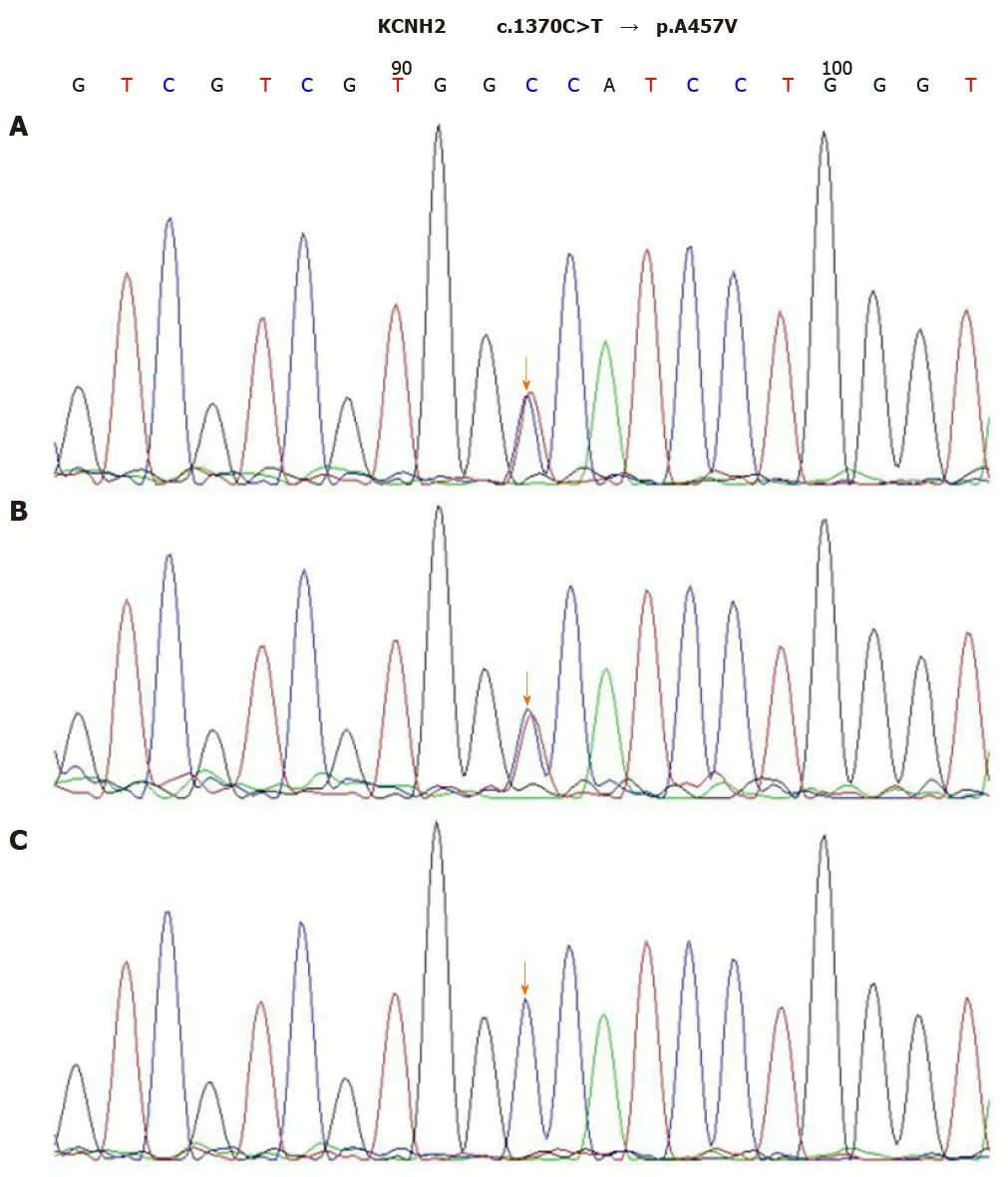
Figure 2 Sequence chromatograms of KCNH2. A and B: The patient and her father had a heterozygous mutation of c.1370C>T (orange arrow); C: Her mother had no relevant genetic mutations detected (orange arrow).
DISCUSSION
TdP is a rare form of fatal arrhythmia usually associated with prolonged QT interval syndrome. Here, we present a case of AOSD in a 30-year old female patient showing long QTc interval complicated with TdP. The key question that we attempted to address here is whether TdP could be induced by the involvement of myocardial dysfunction related to AOSD or the LQTS.
AOSD is a systemic inflammatory disorder presenting daily abruptly spiking fevers,leukocytosis, evanescent rash, and arthritis. It is a rare disease of unknown etiology,and its clinical presentation varies greatly from patient to patient. The diagnosis is mostly clinical and not based upon serology. The Yamaguchi criteria are the most popular of several proposed clinical diagnostic criteria for AOSD[7]. Because of the serosal involvement, cardiac manifestations in AOSD are usually in the form of pericarditis, while myocarditis is rare[5]. We confirmed that the patient in this report met Yamaguchi criteria for AOSD but had no clinical evidence of cardiac involvement otherwise because of her normal results of myocardial enzyme tests, ECG, and CMRI examinations. Therefore, LQTS was speculated to possibly cause the TdP onset.
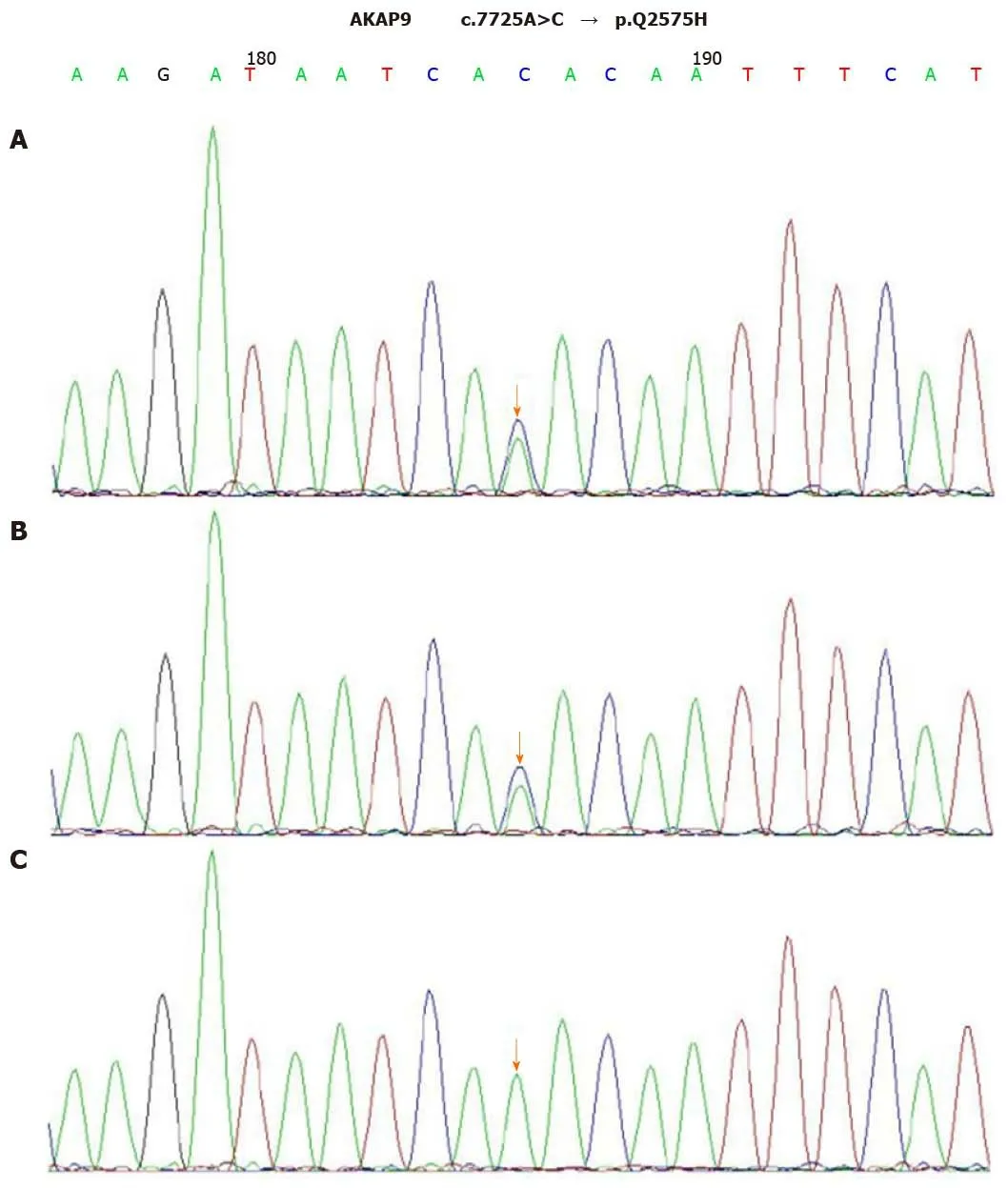
Figure 3 Sequence chromatograms of AKAP9. A and B: The patient and her father had a heterozygous mutation of c.7725A>C (orange arrow); C: Her mother had no relevant genetic mutations detected (orange arrow).
LQTS, including cLQTS or aLQTS, is characterized by QT prolongation and T-wave abnormalities due to delayed ventricular repolarization. cLQTS is clinically uncommon, estimated as 1:2500. It has been reported that this is a result of mutations in genes encoding or regulating cardiac ion channels. Clinically, cLQTS patients often first present recurring episodes of syncope and/or seizure, then subsequent ECG reveals prolonged QT interval. Clinical scoring systems in combination with genetic testing can be used to assist the precise diagnosis of cLQTS, particularly in cases of borderline QT intervals or equivocal clinical history. In this report, the patient had a normal baseline QT interval, and the cumulative LQTS risk score calculated using the 2011 LQTS diagnostic criteria was 1 point, which indicated a low probability of cLQTS[8].
The common causes of aLQTS are side-reactions of QT-prolonging drugs, electrolyte disturbances, and bradyarrhythmias. Although rarely, some non-cardiac drugs such as antibiotics have recently been found to cause QT prolongation and/or TdP onset. In 2010 and 2019, two studies using the United States Food and Drug Administration Adverse Event Reporting System indicated macrolides, fluoroquinolones, and linezolid as TdP-inducing agents[9,10]. Macrolides, fluoroquinolones, and the azole antifungal agents are well-known to prolong QTviablockade of the rapidly activating delayed rectifier potassium channel (hERG/IKr channel). The combined therapy of these QT-prolonging drugs, in this case, could increase the risk of TdP pathogenesis.There is increasing awareness of the impact of gender and gonadal steroids on human cardiac rhythm and episodes of arrhythmias. For example, women have been shown to be more susceptible to develop TdP when using drugs that alter ventricular repolarization[11-13]. In addition, both clinical and experimental data implicated gonadal steroids (estradiol and testosterone) as determinants of gender-related differences in repolarization. Notably, estradiol is considered to promote QTc lengthening, while progesterone and testosterone shorten QTc. Prolonged repolarization, transmural dispersion, and early after-depolarizations are critical factors for the induction of TdP.Together, these factors can establish pathogenically preferred conditions predisposing females a greater risk for proarrhythmic effects of drugs[14,15]. It is now becoming evident that sex hormones play an important role in cardiac repolarization and the maintenance of QT interval.
Another striking feature of this case is the combination of systemic inflammatory activation and episodic high fever upon long QTc and TdP onset. An emerging body of evidence indicates systemic inflammation as a potential cause of aLQTSviacytokine-mediated changes in cardiomyocyte ion channels[16,17]. Many basic experiments demonstrated that induction of inflammatory cytokines, mainly tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6), can directly cause the dysfunction of several cardiac ion channels, particularly K+channels, leading to ventricular action potential duration (APD) prolongation. TNF-α/TNFR1 system can impair hERG/IKr function mainly by stimulating reactive oxygen species production in cardiomyocytes[18]. In excised papillary muscles, IL-1 modified electrical propertiesvialipid second messengers generated by cyclo-oxygenase and lipoxygenase pathways[19]. Aromolaranet al[20]reported that IL-6 inhibition of IKr and the resulting prolongation of APD were mediatedviaIL-6R and Janus kinase pathway activation, and that formed the basis for the observed clinical QT interval prolongation. In patients with elevated CRP levels under different inflammatory conditions, QTc prolongation remained a common pathological hallmark.Interestingly, CRP reduction was associated with significant QTc shortening, which might correlate with the decrease of IL-6 level[16]. Unfortunately, cytokine levels were not measured in this case; however, significantly elevated CRP levels reflected systemic inflammatory activation. Besides these direct effects, systemic inflammation might also indirectly favor LQTS/TdP-axis by several additional mechanisms,including the induction of episodic fever and related temperature-mediated changes in the biophysical properties of cardiac ion channels, particularly the hERG-channel. It has been shown that febrile temperature facilitates the development of LQTS by enhancing hERG degradation through altered K+dependence[21,22]. Fever is more likely to induce LQTS in conditions where hERG function is partially compromised, such as hypokalemia that may promote proarrhythmic potential of IKr blockers and other conditions associated with prolonged ventricular repolarization[23].
Genetic channelopathies involving pathogenic mutations in 17 different genes have been currently identified in clinically diagnosed LQTS. LQTS-causing mutations have a prevalence ratio of approximately 1:2000 in apparently healthy live births. Mutations can involve genes encoding the channel-protein itself or ion channel regulatory proteins, resulting in either loss-of-function of one of the K+currents or gain-offunction of the Na+or Ca++currents. LQT1 (KCNQ1, 30%-35%), LQT2 (KCNH2, 20%-25%), and LQT3 (SCN5A, 5%-10%) represent most of genotype-positive cases[24,25]. A number of lines of evidence suggest that genomic variants may contribute to variable susceptibility to drug-induced TdP onset. An international collaborative study showed that one-third of the patients with aLQTS carry cLQTS-related gene mutations,especially those on KCNH2 being more common[5]. The case in our report had compound heterozygous mutations inKCNH2andAKAP9genes. Notably, IKr and IKs are two important K+currents participating in ventricular repolarization.KCNH2mutations produce defective hERG protein, resulting in a decrease in IKr activity,whileAKAP9mutations with a low frequency (< 1%) disrupt its interaction with KCNQ1, reducing cAMP-stimulated phosphorylation of KCNQ1 and abolishing IKs upregulation. Therefore, we conclude that TdP with long QTc, in this case, could be associated with the patient’s genetic background and the acquired triggering effects from sex, antibiotics, high-grade systemic inflammation, and fever.
Now, a novel and more comprehensive classification of cardiac channelopathies has been proposed, distinguishing the “classic” inherited forms, related to genetic mutations, from the acquired forms, including drug-induced and more recently recognized inflammatory and fever-induced cardiac channelopathies[5]. Furthermore,multiple QT-prolonging factors are also concomitantly required to significantly disrupt ventricular repolarization. Besides specific inherited or acquired channelopathies, drugs, and inflammation, physiological (e.g., age, sex, common polymorphisms, autonomic changes, and exercise) or pathological conditions (e.g.,electrolyte imbalances, heart diseases) may be superimposed in an intricate and often unpredictable scenario inducing TdP[26].
CONCLUSION
This case study highlights the risk of systemic inflammatory activation and antibioticinduced TdP/LQTS onset. Genetic analysis should be considered to identify individuals at high risk of developing TdP.
 World Journal of Clinical Cases2021年12期
World Journal of Clinical Cases2021年12期
- World Journal of Clinical Cases的其它文章
- Standardization of critical care management of non-critically ill patients with COVID-19
- Mediastinal lymphadenopathy in COVID-19: A review of literature
- Polycystic ovary syndrome: Pathways and mechanisms for possible increased susceptibility to COVID-19
- Circulating tumor cells with epithelial-mesenchymal transition markers as potential biomarkers for the diagnosis of lung cancer
- Clinicopathological features of superficial CD34-positive fibroblastic tumor
- Application of a rapid exchange extension catheter technique in type B2/C nonocclusive coronary intervention via a transradial approach