
A Novel Human Antibody, HF, against HER2/erb-B2 Obtained by a Computer-Aided Antibody Design Method
2021-04-24 03:17ChunxiaQiaoMingLvXinyingLiXiaolingLangShouqinLvMianLongYanLiShushngGngZhouLinBifnShnJiannanFng
Engineering 2021年11期
Chunxia Qiao, Ming Lv, Xinying Li, Xiaoling Lang, Shouqin Lv, Mian Long, Yan Li,Shushng Gng, Zhou Lin, Bifn Shn,*, Jiannan Fng*
a State Key Laboratory of Toxicology and Medical Countermeasures, Institute of Pharmacology and Toxicology, Beijing 100850, China
b Beijing Key Laboratory of Therapeutic Gene Engineering Antibody, Beijing 100850, China
c Institute of Military Cognitive and Brain Sciences, Beijing 100850, China
d Beijing Mabworks Biotech Co. Ltd., Beijing 100176, China
e Institute of Mechanics, Chinese Academy of Sciences, Beijing 100190, China
Keywords:HER2/erb-B2 Human antibody Computer-aided design
ABSTRACT Fully human antibodies have minimal immunogenicity and safety profiles. At present, most potential antibody drugs in clinical trials are humanized or fully human. Human antibodies are mostly generated using the phage display method (in vitro) or by transgenic mice (in vivo); other methods include B lymphocyte immortalization,human-human hybridoma,and single-cell polymerase chain reaction.Here,we describe a structure-based computer-aided de novo design technology for human antibody generation.Based on the complex structure of human epidermal growth factor receptor 2(HER2)/Herceptin,we first designed six short peptides targeting the potential epitope of HER2 recognized by Herceptin.Next,these peptides were set as complementarity determining regions in a suitable immunoglobulin frame, giving birth to a novel anti-HER2 antibody named ‘‘HF,” which possessed higher affinity and more effective anti-tumor activity than Herceptin. Our work offers a useful tool for the quick design and selection of novel human antibodies for basic mechanical research as well as for imaging and clinical applications in immune-related diseases, such as cancer and infectious diseases.
1. Introduction
Monoclonal antibodies (mAbs) have been an important therapeutic choice for many diseases, and are expected to play a significant role in relative disease treatment in future [1,2]. The development of antibody-based drugs began with mouse hybridomas; however, early attempts to make mouse antibodies as therapeutic drugs were completely unsuccessful, mainly because of the immunologic reaction and inefficient effector functions. Novel technologies for the preparation of chimeric or humanized mAbs have alleviated these problems. Chimeric/humanized mAbs with similar or stronger antigen binding than the parental murine antibody have been demonstrated to have weaker immunogenicity and lower cellular toxicity,providing better therapeutic efficacy.Since 1990s,antibodies have been viewed as a new and important type of drug for clinical use in chronic inflammatory diseases, oncology, infectious diseases, transplantation,and cardiovascular medicine.However,developing a chimerized or humanized antibody is challenging. Chimerization and humanization can both lead to reduced affinity and/or biological activity, and ensuring the consistency of the binding affinity and biological activity is time consuming and costly. Furthermore, the remaining non-human portions of chimeric and humanized mAbs can still cause an immunogenic reaction,especially after 3-5 years.
New technologies for the preparation of human mAbs are now beginning to decrease the need for chimeric/humanized antibodies. Human antibodies theoretically have minimal immunogenicity, along with favorable safety profiles. Most antibodies currently entering clinical trials are fully human, and this is perceived as the development trend of antibody agents.
Several types of technologies are used to produce human antibodies: phage display, which can establish human antibody libraries; human-human hybridoma; transgenic mice; B cell immortalization and cloning; and single-cell polymerase chain reaction (PCR). Most human antibodies currently under development are generated from phage display (in vitro) or from transgenic mice (in vivo) containing human antibody genes. Through phage display technology, a special human antibody can be screened out, avoiding the need for immunizing animals. Several fully human antibody drugs obtained from phage display are at different stages of clinical trials. In January 2003, Humira (adalimumab) entered the market as the first antibody drug screened from the phage library launched by Abbott. Humira has been approved for the treatment of multiple diseases, including rheumatoid arthritis, plaque psoriasis, and Crohn’s disease. However, the phage antibodies that were isolated in the first round of trials for Humira typically showed a low binding affinity, so additional research is necessary in future to mature their affinity. The affinity of human antibodies obtained from transgenic mice is often high enough, indicating that in vivo affinity maturation is integral to a reinforced immune response.Normally,the high affinity obviates the need for subsequent in vitro affinity maturation or for potency enhancement of mAbs using other technologies. In 2006, panitumumab, which inhibits the epidermal growth factor receptor signal pathway, became the first mAb drug derived from a transgenic mice platform.At present,a series of human antibodies generated from transgenic mice have been reported and are good candidates to enter clinical trials.
In many recent clinical trials,antibody candidates with low toxicity and high efficacy have become the focus of expectations for accelerating the development of next-generation drugs.However,it is quite time consuming to obtain therapeutic antibodies.Efforts to enhance antibodies’pharmaceutical characteristics—such as affinity[3,4], effector bio-function [5], and biophysical and biochemical stability[6,7]—have been effective through structure-based design.
Computer-aided homology modeling has contributed a great deal to the development of therapeutic antibodies, especially in the early stages of humanizing antibodies. The overall potential of computational methods has not yet been well defined,and these methods are not well developed, in contrast to the computational methods used in small-molecule drug discovery.Given the current depth of understanding of antibodies’ mechanisms and the new level of structural resolution available,we think it may be possible to design novel antibody molecules using computer-aided molecular design methods. In our previous work, we created a series of functional antibody-mimicking molecules including peptide-Fc fusion proteins and single-domain antibodies [8-11].
In this paper,we describe a novel fully human antibody targeting human epidermal growth factor receptor 2 (HER2/erb-B2),achieved by means of structure-based computer-aided de novo design. HER2/erb-B2 is over-expressed in approximately 20%-30%of invasive breast cancers and has been identified as an important marker against cancer [12]. Several antibodies that target HER2, such as Herceptin (trastuzumab), show effective antitumor activity both in vitro and in vivo in patients with breast cancers [13,14]. X-ray crystallography and molecular modeling have provided molecular details of the interaction between HER2 and Herceptin [15]. Based on this information, the potential epitopes of HER2 identified by Herceptin were determined. Using the antibody de novo design method that we had previously established,a novel anti-HER2 human mAb named ‘‘HF,” which possessed favorable anti-tumor activity, was obtained.
2. Materials and methods
2.1. Reagents
HER2 protein(catalogue number:10818-H08H)was purchased from Sino Biological Inc., China; Human IgG Isotype Control (catalogue number:02-7102)was from Invitrogen,USA;phycoerythrin(PE) conjugated goat anti-human polyclonal antibody (PE_GAH)(catalogue number:12-4998-82)was purchased from eBiosciences,USA; Dulbecco’s modified Eagle’s medium (DMEM) (catalogue number:11965-092)and fetal bovine serum(FBS)(catalogue number: 10438-034) were from Thermo Fisher, USA; DELFIA EuTDA Cytotoxicity Reagents(catalogue number:AD0116)were purchased from Perkin Elmer,USA,and all other reagents were obtained from a commercial source at analytical grade.
2.2. Cell lines
Human breast cancer cell lines SKBR3 (ATCC®HTB-30), BT474(ATCC®HTB-20), MCF-7 (ATCC®HTB-22), T47D (ATCC®HTB-133),human ovarian cancer cell line SKOV3 (ATCC®HTB-77), human hepatoellular carcinomas (HepG2) (ATCC®HTB-8065), and human embryonic kidney epithelial cells 293T (ATCC®CRL-3216) were purchased from American Type Culture Collection (ATCC, USA).Human peripheral blood mononuclear cells(PBMCs)were distilled from whole blood using Ficoll-Paque PLUS(catalogue number:17-1440-02,GE Healthcare,USA).Cells were cultured in DMEM(catalogue number: 11995-500, Gibco, USA) supplemented with 10%heat-inactivated FBS (catalogue number: 26010-074, Gibco, USA),100 units·mL-1of penicillin, and 100 units·mL-1of streptomycin(catalogue number: 15140-122, Gibco, USA) in 5% CO2at 37 °C.Cells were tested to confirm that they did not contain mycoplasma contamination using a LookOut Mycoplasma PCR detection kit(catalogue number: MP0035, Sigma-Aldrich, USA).
2.3. Computer-aided humanization and epitope prediction
Three-dimensional (3D) structures of HER2 or antibodies were constructed by means of the computer-aided homology modeling method using InsightII (Version 2005, Molecular Simulations,Inc., USA). Potential epitopes were analyzed by the 3D complex structure of HER2/antibodies modeled by molecular docking and optimized using the modules Discover and CHARMM in InsightII.The non-bonded cutoff was 10 Å, and non-bonded parameters and atomic charges were taken as defaults. A distance-dependent dielectric constant was used in the in vacuo calculations.The model was minimized using the steepest descent(2000 steps)and conjugate gradient (5000 steps) methods, respectively.
Based on the epitope,six peptides were designed and set as the six complementarity determining regions (CDRs) in a suitable frame-work region (FR), giving birth to a novel antibody HF. Then the theoretical structure of the HER2/HF complex was analyzed in terms of the potential epitopes, affinity, and so forth.
2.4. Antibody-dependent cell cytotoxicity
The cytotoxicity assay activities of the antibodies were tested using DELFIA EuTDA Cytotoxicity Reagents (PerkinElmer, USA)according to the manufacturer’s instructions. In brief, the target cells (i.e., tumor cells) were incubated with fluorescenceenhancing ligand, then incubated with the diluted antibodies at 37 °C for 1 h, followed by the addition of the effector cells (i.e.,human PBMCs). The effector-to-target ratio was 50:1. After an additional incubation at 37°C for 4 h,the supernatant fluorescence was measured using a time-resolved fluorometer instrument.Maximum release was determined using cell samples lysed in 0.2%Triton X-100 (Gibco, USA). The lysis percentage in each mAb-treated sample was calculated by the following formula:

2.5. Biacore
The commercial antigen HER2 protein was set as the fluid phase, and Herceptin was set as the positive control. The solvent phase (i.e., sample buffer) was set as the negative control. The background was deducted according to the V-baseline in order to obtain ideal kinetic curves and calculate the dissociation constant(KD) value. Both HF and Herceptin had a KDvalue of 148 kDa for calculating the molar concentration.
2.6. Flow cytometry
Experiments were performed on ice. Cells were washed and then incubated with a series of diluted mAbs or immunoglobulin G (IgG) isotype control. After washing and incubation with PE_GAH, cell samples were determined using a BD FACSCalibur(BD Biosciences, USA), and the fluorescence density was analyzed by BD CellQuest Pro software (BD Biosciences, USA).
In the epitope identification assays, using the alanine replacement method, a series of HER2 mutants were synthesized and expressed on the 293T cell surface.Next,cells were incubated with HF as the primary antibody and then with PE_GAH as the secondary antibody for detection.
2.7. In vivo assays in mice
A total of 18 8-10-week-old female BALB/c nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd., China. The mice were housed in filter-top cages (three mice per cage) in a specified pathogen free (SPF)-level facility,and fed with sterilized water and food. During the in vivo assays,we followed the Animal Research: Reporting of In Vivo Experiments (ARRIVE)) guidelines for the use of mice. We observed animal ethics according to the 3R principles (namely, replacement,reduction, and refinement), and the use or treatment of the mice was in strict agreement with the guidelines for research animal usage and was approved by the Animal Ethics Committee of the Beijing Institute of Pharmacology and Toxicology. The mice were checked for discomfort and general appearance. After the study,the mice were sacrificed using anesthesia followed by the cervical dislocation procedure;we used deep-freezing to confirm the death.
For the xenograft model, a number of 2 × 106tumor cells per mouse were subcutaneously (s.c.) injected. After a week, the mice were randomly divided into three groups (the number of animals per group was 6), administered via intravenous (i.v.) with human IgG isotype control or mAbs, and observed twice a week for four weeks after the administration of the antibody.On day 28,the mice were observed or sacrificed to separate the organs for imaging.The mice were first anesthetized by the intraperitoneal(i.p.)injection of 80 mg·kg-1ketamine, 20 mg·kg-1xylazine, and 0.6 mg·kg-1atropine mixture before antibody administration. Each mouse was imaged using a VersArray 1300B device detector (Roper Scientific,the Netherlands),and photons were counted after 5 min exposure.The experiments were performed twice. MetaVue software(Molecular Devices,USA)was used for data analysis.
2.8. Statistical analysis
All experiments were done at least two times. Data are expressed as mean±standard deviation(SD).Pair-wise differences between several groups were compared. Statistical analysis was performed by Student’s t test or by repeated measures of oneway analysis of variance (ANOVA). Treatments were taken as statistically significant when the P values were less than 0.05.
2.9. Ethical approval
The use or treatment of mice was in strict agreement with the guidelines for research animal usage and was approved by the Animal Ethics Committee of the Beijing Institute of Pharmacology and Toxicology.
3. Results
3.1. Novel human antibody HF designed by a computer-aided design method
Our de novo human antibody design scheme consists of five main steps. ①The functional epitopes of the antigen (i.e., HER2)were determined according to the 3D structures of the antigen and antigen-antibody complex (i.e., HER2-Herceptin) and analyzed using biological site-directed mutagenesis. ②Based on the surface physicochemical properties of the functional epitopes,considering the interaction modes (e.g., intermolecular hydrogen bonds, electrostatic and Van der Waals interactions) between the potential key residues and the epitopes, the key amino acids were screened and oriented. Suitable distances and angles were calculated. To ensure a suitable position for the potential key residues,glycine,serine,and alanine were introduced as the linker.③According to the homologies of the CDRs contrasting with the variable regions of the heavy chain of immunoglobulin (IGHVs)/variable regions of the kappa chain of immunoglobulin (IGKVs)in the variable regions of immunoglobulin (IGV) library, we chose the human antibody heavy chain variable region IGHV3-66 and the light chain variable region IGKV1-39 as scaffolding to optimize and display the screened key amino acid, and then to realize virtual affinity maturation. Based on the potential 3D scaffold structures(i.e., the framework regions of IGHV3-66 and IGKV1-39), the suitable peptides designed in Step 2 were replaced in the six CDRs.④Based on the designed antibody sequences, 3D structures of the variable fragment (Fv) were constructed, including the Fv of the heavy chain (HFv) and the light chain (LFv). The orientation and the conformation of the Fv fragments were analyzed one by one, and stable conformations of the Fv fragments were kept to study deeply. ⑤The 3D structures of the novel antibody variable domains LFv and HFv were constructed using a computer-aided homology modeling method. The structural complementarities between LFv and HFv were evaluated, and 3D structures of the Fv fragments were constructed using a molecular docking method.Furthermore,HER2-Fv complex models were constructed using the docking method and optimized with molecular dynamics. Using the binding energy and stable energy as criteria,the designed novel antibody Fv fragments with the most stable and lowest binding energy with HER2 were chosen for testing.
To define the molecular basis for the functional domain of the protein HER2, we analyzed the 3D crystal complex of HER2-Herceptin (Protein Data Bank code: 1n8z) and predicted the key identified domains of HER2(Fig.1(a)).Making use of methods such as solvent accessible surface area calculation and surface electrostatic potential distribution to understand the interaction between HER2 and Herceptin [15], we identified the key residues in the C-terminus of the human HER2 extracellular domain (Fig. 1(b)).Residues Y560, K561, D562, and F565 (herein named as D1) were identified by Herceptin. As residues K585 and I583 (herein named as D2) possessed better accessibility and a more positive electrostatic potential than D1, they were first predicted to be the potential key domain. The domain named as D3, which included D552,Q553, and E550, was used as a comparison in the research. The experimental results showed that the theoretical result was rational (Fig. 1(c)). Since residues 582-585 were near the membrane and close to important epitopes (i.e., from 560-565 of HER2 identified by Herceptin), we considered these nearmembrane residues to be another key domain of HER2.

Fig. 1. 3D structure of the extracellular domain of human HER2 and epitopes prediction. (a) 3D theoretical ribbon structure of HER2 based on the computeraided homology modeling method according to the crystal structure of HER2;(b) potential key amino acid residues in the HER2 C-terminus; (c) orientation of potential key amino acid residues in the HER2 C-terminus.
Residues K561 and K585 in HER2 formed a positive potential location,while residue D562 formed a negative potential location;residues F565 and I583 formed a hydrophobic core, whereas residues D562, K585, K561, and Y560 formed a hydrophilic core. Furthermore, the side chain aromatic ring in residues F565 and Y560 formed a π-π interaction.
We selected and oriented the key residues using a computeraided molecular design method, which took into consideration the physicochemical properties of the amino acids’ potential intermolecular interactions (e.g., hydrophobic interactions, π-π sandwich complexes, hydrogen bonds, salt bridges, polar interactions) and the intermolecular binding distance. Based on the spatial conformations of the two determined potential domains (i.e.,from residues 560 to 565 and from 582 to 585 of HER2),the possible key residues were predicted using computer-aided virtual screening. After studying the stability and distribution of the human antibody variable region,the framework of the human antibody heavy chain variable region IGHV3-66 and light chain variable region IGKV1-39 were chosen as the scaffold to display the suitable key amino acids in order to construct a novel antibody.We established the topology structure of the chosen framework of the heavy chain variable region (Fig. 2(a)) and light chain variable region (Fig. 2(b)). According to the chosen scaffold and key residues, based on computer-aided homology modeling and the molecular docking method, the most suitable human antibody was screened.
Using computer-aided homology modeling and molecular mechanics optimization, 3D structures of the novel human antibody were constructed. Based on the stable energy of the Fv, and the interaction between the variable domain of the heavy chain(VH)and the variable domain of the light chain(VL),the conformation and orientation of the selected key residues in the CDRs were analyzed and suitable antibodies were selected.
Based on the structure of HER2 and its functional antibody Herceptin, the structures of HER2 (Fig. 1(a)) and the selected human antibodies were modeled using the computer-aided molecular docking method. The interaction energy, intermolecular hydrogen bonds, and hydrophobic interactions were analyzed. Finally, we chose the antibody named HF,which possesses more stable energy,better binding,and better specificity for the key domain mentioned above.
The 3D structure of HF is shown in Fig. 3(a). The hydrophilic core formed by the four CDRs (i.e., HCDR1, HCDR2, LCDR1, and LCDR2) contained the CDR3s (i.e., HCDR3 and LCDR3). Residues His91 and Tyr92 in the LCDR3, Phe27 in the HCDR1, and Phe104 in the HCDR3 formed the hydrophobic core and possessed an aromatic property.Residues Gln27 in the LCDR1,Asn28 in the HCDR1,Asn55 in the HCDR2,and Asp102 in the HCDR3 formed a polar and hydrophilic pocket to contain the hydrophobic core.
The 3D complex structure of HER2 and the novel antibody HF was constructed as shown in Fig.3(b).Comparing the crystal structure of HER2 and Herceptin, the binding mode between HF and HER2 was closer than that between Herceptin and HER2. Furthermore, the novel antibody HF identified the novel epitope D2 in addition to the epitope D1, which was specifically identified by Herceptin.
3.2. Construction and identification of the novel antibody HF
We used reverse translation to design the sequence of the variable region of the novel anti-HER2 antibody HF (Chinese patent ZL200910131355.3) for mammalian cell expression. The HF VH and VL gene segments were cloned by means of overlapping PCR,and the antibody HF was constructed by inserting the variable region into an expression vector containing IgG1 constant domains. The expression levels of the HF antibody were 5-10 μg·mL-1,as judged by sandwich enzyme-linked immunosorbent assay using immobilized goat anti-human IgG (data not shown). The antibody was affinity purified using protein A chromatography to show a single band with the expected molecular weight (MW) of approximately 150 kDa by Coomassie bluestained sodium dodecyl sulfate-polyacrylamide gel electrophoresis.Electrophoresis assays (reduced) showed two bands, which were consistent with the expected MW of single heavy(50 kDa)and light(25 kDa) chains (data not shown). N-terminal sequencing and matrix-assisted laser desorption/ionization time-of-flight peptide mass fingerprinting dentification gave the expected sequence for the light and heavy chains(Chinese patent ZL 200910131355.3).

Fig. 2. Design of the novel anti-HER2 antibody HF. (a) Topology structure of the chosen framework of the heavy chain variable region, analyzed using the Collier-de-Perles tool.(i)One-layer topology structure;(ii)two-layer topology structure.(b)Topology structure of the chosen framework of the light chain variable region,analyzed using the Collier-de-Perles tool. (i) One-layer topology structure; (ii) two-layer topology structure.
The antigen-recognition capabilities of HF were assessed by flow cytometry analysis.Several human tumor cell lines were incubated with the antibody. The results indicated that HF could bind to HER2 over expression cell lines only, such as SKOV3, SKBR3,MCF7, and T47D (Fig. 4(a)). To further identify the specificity of HF to bind HER2, we fused the gene encoding HER2 extracellular domain (ECD) and expressed it on the membrane of 293T cells, with enhanced green fluorescent protein (EGFP) transfected cells set as the negative cell control. The EGFP-positive cells were sorted to show the expression of HER2. When the cells were exposed to HF,we analyzed the cells with both a green fluorescent protein (GFP)+ (that was HER2+) and PE_GAH positive signal to show HF binding. Both the flow cytometry assay (Fig. 4(b)) and the fluorescent microscopy detection (Fig. 4(c)) results showed that the 293T cells that over-express HER2 can be specially identified, while the cells that only express EGFP cannot. The data support the idea that HF specifically recognized HER2.
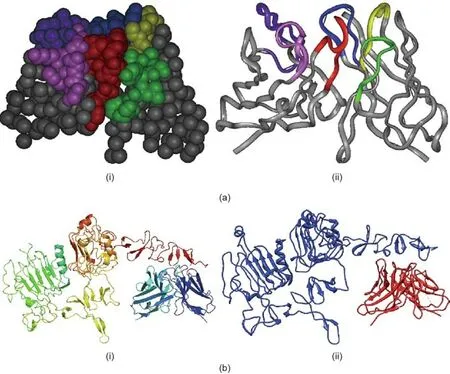
Fig.3. 3D complex structure of HER2 and HF.(a)Rationally chosen CDR residues based on the topology structure of the antibody variable region framework.The top-down view of the CDR binding loops of HF as (i) a space-filling model and (ii) a ribbon structure are shown, highlighting the residues chosen for optimization in the heavy chain CDR-H1 (yellow), CDR-H2 (green), and CDR-H3 (blue), and the light chain CDR-L1 (pink), CDR-L2 (magenta), and CDR-L3 (red). Kabat numbering is used in this and subsequent figures. (b) 3D structure of HER2 to bind to Herceptin ((i) the crystal structure of HER2-Herceptin) and HF ((ii) the modeling structure of HER2-HF).
HER2 ECD contains two domains: an N-terminal domain and a C-terminal domain. These domains mediate different biological functions. When we designed the HF antibody, the epitope was theoretically contained in the C-terminus.To verify the rationality of the design and determine the target domain of HF, two truncated mutants, HER2-C and HER2-N, were constructed. The data from fluorescent microscopy observation and fluorescence activating cell sorter (FACS) assays suggested that, similar to Herceptin,HF bound to the HER2 C-terminal domain, which was in accordance with the preliminary design concept (Fig. 4(d)).
3.3. The epitope of HF was superimposed but different from that of Herceptin
Based on X-ray crystallography data and molecular models of HER2, three potential epitopes were predicted: 550/552/553,560/561/562/565, and 583/585.According to the X-ray crystallography of the complex that formed between HER2 and Herceptin,the Herceptin epitope was 560/561/562/565,while the HF epitope was a spatial conformation built by 560/561/562/565 and 583/585 based on the molecular structure of the HER2/HF complex.To confirm these predictions,we constructed a series of membrane HER2 mutants through alanine replacement named as:mHER2,mutated at 550/552/553, 560/561/562/565, and 583/585; mHER2-M1,mutated at 550/552/553; mHER2-M2, mutated at 560/561/562/565; and mHER2-M3 mutated at 583/585 (Fig. 5(a)). These mutants were expressed on a 293T cell membrane fused with EGFP. After incubation with Herceptin or HF followed by PE_GAH,the cells were analyzed by FACS and fluorescent microscopy detection. The results indicated that ①mHER2 cannot be identified by Herceptin or HF (Figs. 5(b) and (c)); ②the epitope 550/552/553 does not participate in the interaction between HER2 and Herceptin or HF, as the mutant mHER2-M2 was not identified by Herceptin or HF (Fig.5(d)); and ③mHER2-M3 interacts with Herceptin but not with HF. Taken together, these findings indicated that the epitope of HF was a spatial conformation built by 560/561/562/565 and 583/585, which differed from that of Herceptin.
Furthermore, each of the two antibodies was conjugated with flow cytometry analysis and incubated with SKOV3 cells, while the other antibody (naked) was incorporated as a competition agent. Herceptin could inhibit HF binding to HER2, and the maximum inhibition rate was around 50% when the concentration of Herceptin reached 10 μg·mL-1. Contrarily, HF could not inhibit Herceptin binding activity.This competition profile(Fig.5(e))suggested that the epitope of HF was different from that of Herceptin.
3.4. In vivo anti-tumor activity of HF
HER2-positive cells were incubated with diluted HF to determine their in vitro anti-proliferation activity.Herceptin and IgG were set as controls.HF slightly inhibited HER2-positive cell proliferation.At concentrations greater than or equal to 4 μg·mL-1, it suppressed about 10%of the proliferation of SKOV3 cells(Fig.6(a)).In HER2low+MCF7 cells, almost no anti-proliferation activity was observed(Fig. 6(b)), suggesting that HF directly showed little or no direct anti-tumor activity,which was similar to Herceptin.
However,with human PBMCs as the effector cells,HF efficiently mediated antibody-dependent cell cytotoxicity (ADCC) in the SKOV3, SKBR3, and MCF7 carcinoma cells, which expressed HER2(Fig. 6(c)). Furthermore, the killing activities were proportional to the HER2 expression levels on the cell surface. At the maximal tested concentration (20 μg·mL-1), anti-HER2 antibodies killed 50%-60% of SKOV3 cells, 30%-40% of SKBR3 cells, and 20%-30%of MCF7 cells, with an HER2 expression order of SKOV3 >SKBR3 >MCF7 (Fig. S1 in Appendix A). Interestingly,although the maximal killing rates were similar in HF and Herceptin, HF showed more effective killing activity than Herceptin at a lower concentration. At a concentration of 10 μg·mL-1, HF killed 30.24% ± 5.12% of SKOV3 cells, whereas Herceptin killed 13.73%±1.94%.Similar results were seen in the SKBR3 and MCF7 cells.Aside from the epitope differences, HF might have higher affinity because higher affinity is one of the key factors affecting ADCC activity [16]. The relative affinity of these anti-HER2 antibodies was measured by micropipette adhesion frequency assay [17].The results indicated that the reverse association rate constant(Kon) of HF was higher than that of Herceptin, while the reverse dissociation rate constant (Koff) of HF was lower than that of Herceptin, which means that the relative affinity of HF was advantageous (Fig. 6(d)).

Fig.4. HF bound to the membrane HER2 antigen in vitro. (a) HF bound to the membrane antigen on HER2-positive cell lines;HF bound to the antigen on HER2-transfected 293T cells by(b)flow cytometry analysis and(c)fluorescent microscopy detection.(d)Both Herceptin and HF bound to the HER2 C-terminal domain but not N-terminal.SSC:side scattering; GFP: green fluorescent protein; FITC: fluorescein isothiocyanate; R2: region 2 to select cells for flow cytometry analysis; EGFP: enhanced green fluorescent protein; HER2-C: HER2 C-terminal domain; HER2-N: HER2 N-terminal domain.
3.5. In vivo activity of HF in a xenograft mice model
To assess whether the novel antibody HF inhibits the proliferation of berried solid tumors in vivo, we established a solid tumor model in immune-compromised mice by inoculating them with HER2 over-expressing cancer cells.
BALB/c nude mice were inoculated in the armpit with SKOV3 cells,and HF administration was started eight days after i.v.inoculation with a first dose of 5 mg·kg-1,followed by 2.5 mg·kg-1once a week for four weeks.Tumors were measured with a caliper every three days,and the volume of each tumor was calculated using the formula Volume = (π/6) × (the length of smaller diameter)2× the length of larger diameter. Nonspecific human IgG was used as a negative control agent (the animal number per group was 10,i.v.). Significant tumor growth inhibition was observed by HF (the animal number per group was 10, i.v.) (Fig. 7(a)). At the end of the whole experiment, the average tumor volume in the control mice was as large as (1630 ± 24) mm3, whereas the tumors of HF-treated mice had either disappeared or shrunk to (340 ± 67)mm3on average. Autopsy revealed either no visible tumor or tumors smaller than 400 mm3. All of these statistically significant differences were also observed in Herceptin-treated mice(n = 10, i.v.).
To better define whether there was a dose-response relationship with anti-HER2 treatment, a second SKOV3 xenograft mice experiment was conducted using lower doses of antibody. In this experiment, anti-HER2 antibodies were administered, with the first doses of 1.25, 2.50, or 5.00 mg·kg-1(i.v.), followed by a half dose once a week for four weeks. Marked dose-dependent antitumor activity was observed in both HF-treated and Herceptintreated mice (Fig. 7(b)). Notably, HF revealed more effective antitumor activity than Herceptin at lower doses, although there was no significant difference at 5.00 mg·kg-1. In comparison with the mice treated with the control antibody, the mice treated with 1.25, 2.50, and 5.00 mg·kg-1HF exhibited an average inhibition of tumor growth at 52 days of 49.1% ± 1.6%, 80.8% ± 9.1%, and 78.9% ± 2.4%, respectively. With Herceptin, the inhibition was 22.4%±1.9%,25.8%±4.5%,and 74.2%±11.5%,respectively.Similar results were obtained in solid tumor models in BALB/c nude mice by inoculating another human breast adenocarcinoma cell line,BT474 (Fig. 7(c)).
3.6. In vivo observation of HF accumulation

Fig.5. The epitope of HF was different from that of Herceptin.(a)Three mutants of HER2,namely,mHER2-M1-M3,were designed with the sites 550/552/553,560/561/562/565,or 583/585 replaced with alanine.mHER2 was the triple-mutant.HF and Herceptin had no/very weak capacity to bind mHER2,as shown by(b)fluorescent microscopy and(c)flow cytometry detection.(d)The HER2 583/585 sites seemed to be more important in HF binding than Herceptin,as the mean value of mHER2-M3/HF binding was weaker than that of mHER2-M3/Herceptin.(e)The epitope of HF seemed to be overlapped but not quite the same as the epitope of Herceptin,as Herceptin could inhibit HF binding to HER2 with a maximum inhibition rate of around 50%,while even 50 μg·mL-1 of HF could not inhibit Herceptin’s binding.The nested figures show the experimental concentration of FITC-conjugated antibody (1:25 diluted HF-FITC or 1:125 000 diluted Herceptin-FITC) used in each assay.
In vivo distributions of the HF antibody were observed in SKOV3 xenograft BALB/c nude mice. On day 28 after cell inoculation s.c.,the mice were anesthetized intravenously with 25 mg·kg-1of Cy5.5-labeled HF antibody. The HF gradually penetrated to the tumor and accumulated in the tumor cores, and then spread to the whole tumor area within 4 h. HF molecules still existed in the tumor 48 h after injection (Fig. 8(a)). The mice were then sacrificed,and the tumors and organs were autopsied,which revealed the selective accumulation of HF antibody in the SKOV3-induced tumors. There was no signal in the host vital organs, except for slight accumulation in the liver and kidney(Fig.8(b)),which might be due to metabolism and excretion. To further determine the binding specificity of HF in vivo, we repeated the experiment in nude mice bearing HER2-positive SKOV3 tumors and HER2-negative HepG2 tumors simultaneously. It was found that only the SKOV3 xenograft could accumulate HF, while similar accumulation was not observed in the HepG2 xenografts (Fig. 8(c)). The above data strongly indicated that HF could selectively accumulate in HER2+ solid tumors after i.v. injection.

Fig. 6. In vitro anti-tumor activity of HF. HF showed weak/no inhibition against cell proliferation in (a) HER2-positive SKOV3 or (b) MCF7 cells. (c) HF efficiently mediates ADCC function in HER2-positive SKOV3,SKBR3,and MCF7 carcinoma cells.(d)The affinity of HF was higher than that of Herceptin,as HF possessed a higher Kon as well as a lower Koff constant. ADCC: antibody-dependent cell cytotoxicity; Kon: association rate constant; Koff: dissociation rate constant.

Fig.7. In vivo anti-tumor activity of HF.HF obviously inhibited the tumor growth(a)in a dose-dependent manner and(b)in a SKOV3 xenograft mice model.At a middle or low dosage, HF obviously showed more optimal function than Herceptin. (c) HF inhibited tumor growth in the BT474 xenograft mice model. HF also possessed better inhibitory capacity than Herceptin, especially in the low dosage group. The maximum inhibition rate reached up to ~80%.
4. Discussion
mAb therapy has been facilitated and developed considerably by various technological advances over the past 30 years.Although murine mAbs were discovered first, they failed to become major choices for clinical therapies due to their immunogenicity side effects. Chimeric antibodies showed less immunogenicity but still contained the mouse variable domains of mAbs. Further elimination of mouse sequences was achieved in humanized mAbs,which were followed by fully human therapeutic mAbs. Nowadays,human antibodies have demonstrated broad utility in the clinical therapy of infectious, malignant, and inflammatory diseases.

Fig.8. In vivo targeting and accumulation of HF.(a)Cy5.5-conjugated HF mostly accumulated in the tumor in a dose-dependent manner.(b)HF could be observed mostly in the tumor, with some in the liver or kidney due to metabolism and excretion. (c) HF did not target the HER2-negative HepG2 xenograft after 24 h.
At present, numerous technologies allow the preparation of human antibodies, including phage library technology, transgenic mouse technology, B cell immortalization, human-human hybridoma, and single-cell PCR. However, most human mAb therapies,whether in use or under development, have been derived in nonhuman species, because it is impossible as yet to immortalize and clone human B-lymphocytes. Current methods include singlechain variable fragments (scFvs) encoded by human genes in filamentous phages/yeast, or antibodies screened from transgenic mice that bear human immunoglobulin genes in their chromosomes.
Computational techniques are the focus of current drug design.Structure-based small-molecular design has recently become an important part of the drug discovery process to discover many high-activity molecules[18].Due to the computational complexity of treating large proteins and the relatively scarce information on engineered proteins, computational protein design is challenging[19,20]. Very recently, many achievements have been made in computational protein design; these include the re-design of endonuclease with an internal domain-domain interface [21],the design of a novel fold in a protein [22], the design of a special enzymatic activity in a peri-plasmic binding protein [23], and the alteration of a deoxyribonuclease (DNase)-inhibitor [24]. Computational techniques for antibody molecular design have been demonstrated to be effective in enhancing the antibodies’desirable pharmaceutical characteristics, such as improved affinity [3,25],effector function modulation [5], and enhanced stability [6,7].There has been considerably less usage of computer-aided design techniques in the field of de novo antibody design, however [26].
Here, we presented a design method for generating de novo human antibodies using computer-aided virtual screening and antibody structural information. Furthermore, we characterized the novel antibody HF and compared it with the therapeutic antibody Herceptin.
In principle,the strategy presented here is applicable to any de novo antibody molecule designed on the basis of the spatial conformation of functional epitopes. The proposed approach has several advantages over current techniques. Firstly, it allows effective humanization and affinity maturation. The chosen framework for the design comes from the collected human antibody framework sequence. In addition, the most suitable key residues are screened during the de novo design procedure (Fig. 2). The humanization level of HF was found to be higher than that of Herceptin.Furthermore, the binding ability of HF was better than that of Herceptin(as shown in Fig. 6).
Secondly, the antigen-antibody complex structure can be reliably used in a de novo antibody design procedure. Although computer-aided protein-protein docking methods are improving rapidly,there are still many limitations,especially when theoretical structures are used as the starting points. However, the antibody structures are quite conservative and the binding domains of antigen and antibody are limited to CDRs and epitopes.The information can be helpful for more accurate prediction of the antigen-antibody complex models.With the help of other experimental data such as site mutagenesis,the antigen-antibody docking can be optimized.
To summarize,therapeutic suitability of antibody drugs against cancer, immune-related diseases, infectious diseases, and so forth is expected to drive new innovations.The present method is independent of the commonly used hybridoma technique, antibody humanized technique, and affinity maturation. It is a useful tool for the design of novel human antibodies for basic research as well as for imaging and clinical applications.
Acknowledgments
This work was supported by grants from the National Sciences Fund (31370938 and 81272528). The Fund (81272528) offered experiment material and collected the data for analysis. The Fund(31370938) helped design the study and was helpful in preparing the manuscript.
Compliance with ethics guidelines
Chunxia Qiao, Ming Lv, Xinying Li, Xiaoling Lang, Shouqin Lv,Mian Long, Yan Li, Shusheng Geng, Zhou Lin, Beifen Shen, and Jiannan Feng declare that they have no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.10.024.

- Engineering的其它文章
- PVC Formulation of Anastrepha suspensa Pheromones Suitable for Field Studies
- Handheld Ultrasound Advances Diagnosis
- Nuclear Energy Seeks Revival with Advanced Fuel Options
- Mars Helicopter Exceeds Expectations
- Toward Systemic Thinking in Managing Environmental Risks
- The Experimental Advanced Superconducting Tokamak