
基于加权共表达网络预测结直肠癌的核心基因
2021-04-12 06:49兰燕,张旭
重庆理工大学学报(自然科学) 2021年3期
兰 燕,张 旭
(西南大学 数学与统计学院,重庆 400715)
结直肠癌(colorectal cancer,CRC)是一种发生于直肠或结肠的消化道癌症,是死亡率排名第5位的恶性肿瘤[1]。结直肠癌的死亡率与地区、肿瘤有无发生转移和年龄等多种因素有关。由世界卫生组织国际癌症研究机构的统计结果可知,2008年全球结直肠癌新确诊病例是120万,因结直肠癌致死的人数在癌症死亡总人数中的占比为8%[2]。除了病灶中无腺瘤的新生结直肠癌[3]外,许多结直肠癌发生前常伴有结直肠腺瘤、炎性肠病和家族性腺瘤性息肉病等病变[4]。目前CRC的治疗方法主要有癌胚抗原、CT结肠成像和手术切除等。但由于以上治疗方法在现阶段对结直肠癌的治疗效果不好,故通过研究CRC的发病机制来预测高效的根治结直肠癌的生物标志物十分重要。近年来,许多学者对CRC的发生机制进行了研究,为其诊断和治疗带来了新的突破。Gui等[5]对数据集GSE32323采用差异表达分析、功能富集分析、蛋白互作(PPI)网络分析、药物-基因相互作用分析等方法发现基因MAP2K2,DEPTOR,NEDD9,CCND1,BCAS1和AURKA可能在CRC中起作用。Zhu等[6]对数据集GSE32323和GSE21510筛选公共差异基因(DEGs),对其进行功能富集分析、PPI网络分析和癌症分期分析得到E2F2、SKP2、MYC、CDKN1A、CDKN2B为与CRC相关的核心基因,并采用生存分析和文献法进行验证。Sun等[7]对4个基因表达谱GSE89076、GSE21510、GSE113513、GSE32323采用稳健的秩聚合方法得到差异表达基因,然后根据功能富集分析和MCODE插件的模块 分 析 可 得PPBP、CXCL3、CXCL10、CCL28、CXCL12、INSL5和CXCL11是与CRC相关的核心基因,最后通过生存分析和TCGA数据库验证核心基因。基因表达谱在之前研究中通常用正常样本和肿瘤样本的差异来识别与CRC相关的DEGs。然而,很多研究关注于筛选差异基因,而忽略了表达模式相似的基因之间的高度互连和基因组的复杂网络。由于WGCNA分析可以检测差异相关的基因簇,并通过微阵列数据选择核心基因作为候选的生物标志物,因此本研究对一组与结直肠癌相关的基因表达数据建立加权共表达网络,通过P<0.05筛选出与CRC显著最相关的2个模块作为核心模块,然后用DAVID对其进行GO与KEGG富集分析,再用Cytoscape对核心模块中网络节点按照度排序的前30基因分别进行可视化,得到的60个候选基因,在GEPIA数据库通过生存分析挑出P<0.05的基因,得到PPARG、ACO2、MYC、CCNB2和NUP37为与CRC预后显著相关的核心基因,最后从转录水平、蛋白水平和文献法共同验证选出核心基因。
1 材料和方法
1.1 数据收集和预处理
从基因表达数据库(gene expression omnibus,GEO)下载位于Affymetrix Human Genome U133 Plus 2.0 Array平台的基因表达芯片数据集GSE32323,包括17对结直肠癌组织及其配对的正常组织,共有54 675个探针。对其进行预处理,包括分位数标准化、探针整理,最终以探针水平均值作为基因的最终表达值。由于变异基因通常被认为是背景噪声,通过方差对基因进行筛选,选择方差较大的前25%基因进行后续分析,然后根据皮尔逊相关矩阵和不同样本之间的距离,采用样本聚类的方法来剔除不合格的样本芯片。
1.2 WGCNA分析和功能富集分析
WGCNA是一种基于高通量基因表达谱的算法,是从基因分类的角度利用基因间的相关性来找出与性状显著相关的基因模块。本研究使用WGCNA包构建无尺度网络。首先对表达谱前25%基因建立皮尔逊相关矩阵,接着把皮尔逊相关矩阵转换为邻接矩阵,再计算出平均连接度和无标度指数从1到20的软阈值β,这里β是指基因相关系数的n次幂。然后,把邻接矩阵变换成对应的拓扑重叠矩阵(TOM),基于拓扑重叠矩阵的相异度,采用平均连接层次聚类的方法,从而把相似表达模式的基因划分到同一模块,其中每个共表达模块至少30个基因。设定模块的第1个主成分为特征基因,然后计算出特征基因的皮尔逊相关系数,将特征基因高度相关(Pearson相关系数大于0.75)的模块合并成一个模块,得到划分模块。将不能分到任何模块中的基因放入灰色模块中,在之后的分析中不计。为了验证模块划分的可靠性,先使用R软件给出了WGCNA分析中随机500个基因之间的拓扑重叠邻接热图来展示各模块间的共表达关系。然后通过计算模块-性状相关系数,给出了模块-性状相关系数的热图,先根据模块显著性P<0.05得知模块的特征向量和性状的相关系数具有统计学意义,再选取模块的特征向量和性状的最大相关系数,故通过模块的特征向量与性状的相关系数和模块显著性P筛选出与结直肠癌最相关的2个模块。接着用R软件展示了核心模块中模块成员(MM)与基因显著性(GS)之间相关性的散点图,对核心模块内基因分布进行可视化来证实这些基因适合进一步分析和核心基因的挖掘。为了找到与性状相关模块中基因的生物学功能和潜在的生物学途径,用DAVID数据库对核心模块中基因进行了GO和KEGG富集分析,根据P<0.05分别分析了核心模块中基因的GO富集分析中生物过程(BP)、细胞组成(CC)和分子功能(MF)和KEGG富集分析中按照P值排名前10的通路,这里的P<0.05表示选出的是具有统计学显著差异的通路即显著性富集的通路。
1.3 生存分析筛选核心基因
生存分析是考虑生存时间及其状态的一种统计方法。生存分析主要研究生存现象的响应时间数据及其规律,描述生存过程中的影响因素并对生存结局进行预测[8]。由于核心模块中基因适合进一步分析,故用Cytoscape对选出的核心模块中网络节点按照度排序的前30基因分别进行可视化,然后,在基因表达谱分析(GEPIA)数据库中,对可视化得到的基因进行生存分析,以Logrank P<0.05得到与结直肠癌预后显著相关的核心基因,这里Logrank P<0.05是通过基因的高低表达量存在显著差异来选出具有显著意义的基因。
1.4 GEPIA数据库转录水平和HPA数据库蛋白水平验证核心基因
为了验证这5个核心基因在结直肠癌发生中的重要性,通过GEPIA中的癌症基因组图谱(TCGA)和基因型-组织表达(GTEx)这2个临床样本数据库,分析5个核心基因在结直肠癌样本和正常样本中的mRNA表达水平情况,视满足P<0.05为核心基因存在显著性差异,能分析出这些基因在癌症和正常样本中的上、下调情况。通过人类蛋白质数据库(HPA)中基因在正常和肿瘤组织的蛋白表达量高低从蛋白水平角度对核心基因作验证,褐色越深表示上调。最终把2种方式所得5个核心基因与结直肠癌的关系同WGCNA分析结果进行对比,进一步验证所得核心基因的可信度。
2 结果
数据集GSE32323经预处理后,共留下20 183个基因,选择方差较大的前25%(5 046个基因)进行后续分析。图1(a)为样本聚类树和性状热图,可检测数据集是否有离群值。由于样本GSE800752是离群值,样本聚类排除了样本GSE800752,留下16个正常样本和17个结直肠癌样本。样本聚类的最终结果如图1(a)所示,组内一致性较好,组间差异明显。
2.1 WGCNA分析和功能富集分析
WGCNA根据无标度拟合指数和平均连接度分析确定软阈值β为12,无尺度指数达到0.82时满足无尺度网络构建条件图1(b)。然后把皮尔逊矩阵转化为邻接矩阵,再换成相应的拓扑重叠矩阵得到了10个不同的模块。图1(c)显示了模块内基因和模块颜色可视化情况,上半部分是基因的聚类树,下半部分用不同的颜色显示模块,包括黑色、蓝色、棕色、绿色、灰色、品红、粉色、红色、棕绿色和黄色的模块,分别包含61、1 344、679、101、709、38、53、90、1 806和165个基因。不能进入任何共表达模块中的基因则是灰色模块中的基因。
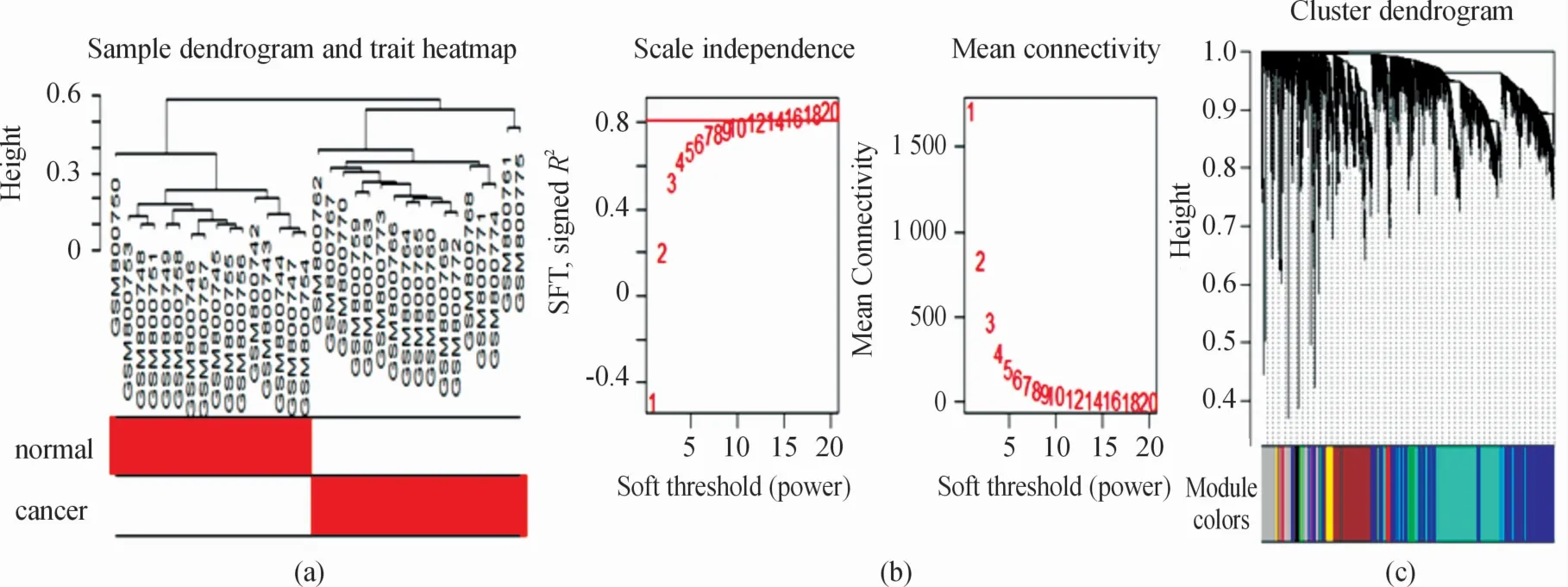
图1 软阈值估计和模块划分示意图
为了验证模块划分的可靠性,图2(a)展示了WGCNA分析中随机500个基因之间的拓扑重叠邻接热图,它是通过计算模块内基因间的连通性得到的聚类结果来展示模块之间共表达关系。横轴和纵轴是不同颜色的模块,热图对角线上中间黄色的亮度代表不同模块的连通性。颜色的深浅表示基因之间的相关性,大部分基因与同一模块的基因相关性较高,与其他模块的基因相关性较低,说明各模块间具有较高的尺度独立性,这一现象说明这些基因的划分是准确的。通过计算模块-特征相关系数,图2(b)显示了模块-性状相关系数的热图,根据模块的特征向量与性状的相关系数和模块相关系数的显著性P值来筛选核心模块。红色为正相关,绿色为负相关,样本的临床特征包括正常和结直肠癌样本,使用P<0.05挑选出了与结直肠癌最相关的核心模块分别为蓝色模块(r=-0.9,p=6e-13)和棕绿色模块(r=0.88,p=1e-11)。从图2(b)中可看出蓝色模块与疾病状态呈负最相关,而棕绿色模块与疾病状态呈正最相关,这一结果表明棕绿色模块在结直肠癌的肿瘤发生中起重要作用,蓝色模块可能起到抗肿瘤的作用。然后用R软件分别展示了2个核心模块中模块成员(MM)与基因显著性(GS)之间相关性的散点图来展示核心模块内基因的分布情况。图2(c)为蓝色模块中MM与GS之间相关性的散点图。图2(d)为棕绿色模块中MM与GS之间相关性的散点图。从散点图顶端得到核心模块中MM与GS之间的相关系数与P值大小,由此可得核心模块MM与GS具有高度相关性,可知与性状高度相关的基因,在与性状相关的模块中也是重要基因,也证明了选取的核心模块中基因适合更深层次的分析和挖掘。

图2 核心模块的识别示意图
为了探索核心模块中基因在CRC中的生物学功能,用DAVID对其进行GO与KEGG分析得到富集分析结果。在蓝色模块中,GO-BP主要富集在细胞增殖、凋亡和迁移的调控、细胞对机械刺激的反应、信号传导等过程。例如细胞增殖的负调控(基因数 =49,p=6.40E-05),凋亡过程负调控(基因数 =52,p=2.75E-04),信号传导(基因数=108,p=6.33E-04)。GO-CC主要富集在膜的组成部分、细胞外泌体、等离子体膜、细胞外间隙等通路。例如膜的组成部分(基因数=400,p=2.26E-04),细胞外泌体(基因数 =287,p=1.21E-14),等离子体膜(基因数 =360,p=4.23E-09)。GO-MF主要富集在蛋白结合、细胞骨架的结构组成等过程。例如细胞骨架的结构组成(基因数=18,p=0.001 256 613),涉及细胞与细胞粘附的钙粘蛋白结合(基因数 =34,p=0.002 706 116)图3(a)。KEGG主要富集在胆汁分泌、代谢通路、TGF-beta信号通路等过程。例如胆汁分泌(基因数 =18,p=7.52E-06),代谢通路(基因数 =128,p=1.41E-05),生物合成的抗生素(基因数 =27,p=0.007 252 557)图3(b)。在棕绿色模块中,GO-BP主要富集在细胞分裂、有丝分裂、DNA复制、细胞增殖等过程。例如细胞分裂(基因数=105,p=4.07E-28),有丝分裂(基因数 =76,p=4.12E-21),DNA复制(基因数 =50,p=5.10E-15),细胞增殖(基因数 =68,p=4.70E-08)。GO-CC主要富集在细胞核、细胞质、核原生质、胞液、纺锤等过程。例如细胞核(基因数 =638,p=2.12E-18),细胞质(基因数 =593,p=5.95E-13),核原生质(基因数=449,p=2.79E-41)。GO-MF主要富集在蛋白结合、ATP结合、酶结合、DNA结合等。例如蛋白结合(基因数=979,p=1.91E-19),ATP结合(基因数 =202,p=1.58E-08),DNA结合(基因数 =194,p=5.71E-04)图3(c)。KEGG主要富集在代谢通路、细胞周期、DNA复制、P53信号通路等。例如代谢通路(基因数 =153,p=6.63E-05),细胞周期(基因数=46,p=1.59E-16),RNA运输(基因数 =34,p=5.24E-05)图3(d)。
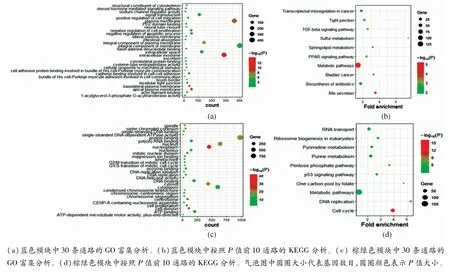
图3 核心模块中基因的功能富集分析示意图
2.2 生存分析筛选核心基因
用Cytoscape对选出的2个核心模块中按照度排序对前30个基因分别进行可视化,得到60个候选基因进行后续研究。然后在GEPIA数据库中通过对这60个基因分别作总生存期生存分析,根据Logrank P<0.05选出与结直肠癌具有显著分析意义的核心基因。从生存分析曲线(图4)可知:红线表示基因高表达,蓝线表示基因低表达,横轴表示为随访时间,纵轴表示成生存率。由图可知:基因的高低表达量开始差异较小,但后期差异较大。本文由Logrank P<0.05得到基因的高低表达量存在显著性预后差异,因此得到结论:蓝色模块中基因PPARG、ACO2与结直肠癌预后显著相关图4(a)~(b),棕绿色模块中基因MYC、CCNB2、NUP37与结直肠癌预后显著相关图4(c)~(e)。
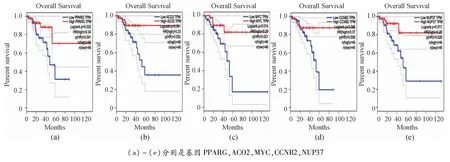
图4 核心基因的总生存期的生存分析曲线
2.3 GEPIA数据库转录水平和HPA数据库蛋白水平验证核心基因
通过GEPIA数据库中包含的TCGA和GTEx数据库,对选出的5个核心基因在转录水平进行验证。2个数据库共包括92个结直肠癌样本以及318个正常样本,图5给出了5个核心基因在直肠癌中肿瘤样本和正常样本的mRNA表达量的高低情况。图6是5个核心基因在正常组织和肿瘤组织中蛋白表达情况,目的是说明这些基因的上调和下调情况,从而验证它们与结直肠癌的相关关系,以证实核心基因选取的准确性。从图5可知:红色表示肿瘤样本,灰色则表示正常样本,横坐标表明5个核心基因在直肠癌中正常样本和癌症样本个数,纵坐标是基因在结直肠癌中的mRNA表达情况,分析可得基因PPARG和ACO2在肿瘤样本中的mRNA表达量低于正常样本,同理可知MYC,CCNB2和NUP37在正常样本中的表达量明显低于肿瘤样本。为了进一步从蛋白水平验证核心基因,给出了图6所示的5个核心基因在HPA数据库中的免疫组化染色分析结果,图6左侧给出了正常组与癌症组的抗体编号和病人编号,可知PPARG和ACO2基因在正常组中上调,而MYC,CCNB2和NUP37在结直肠癌组中上调,故得出蓝色模块中核心基因PPARG和ACO2与结直肠癌负相关,棕绿色模块中的3个核心基因:MYC,CCNB2和NUP37与结直肠癌正相关,这与WGCNA所得结果一致,也证实所得核心基因的可信度。
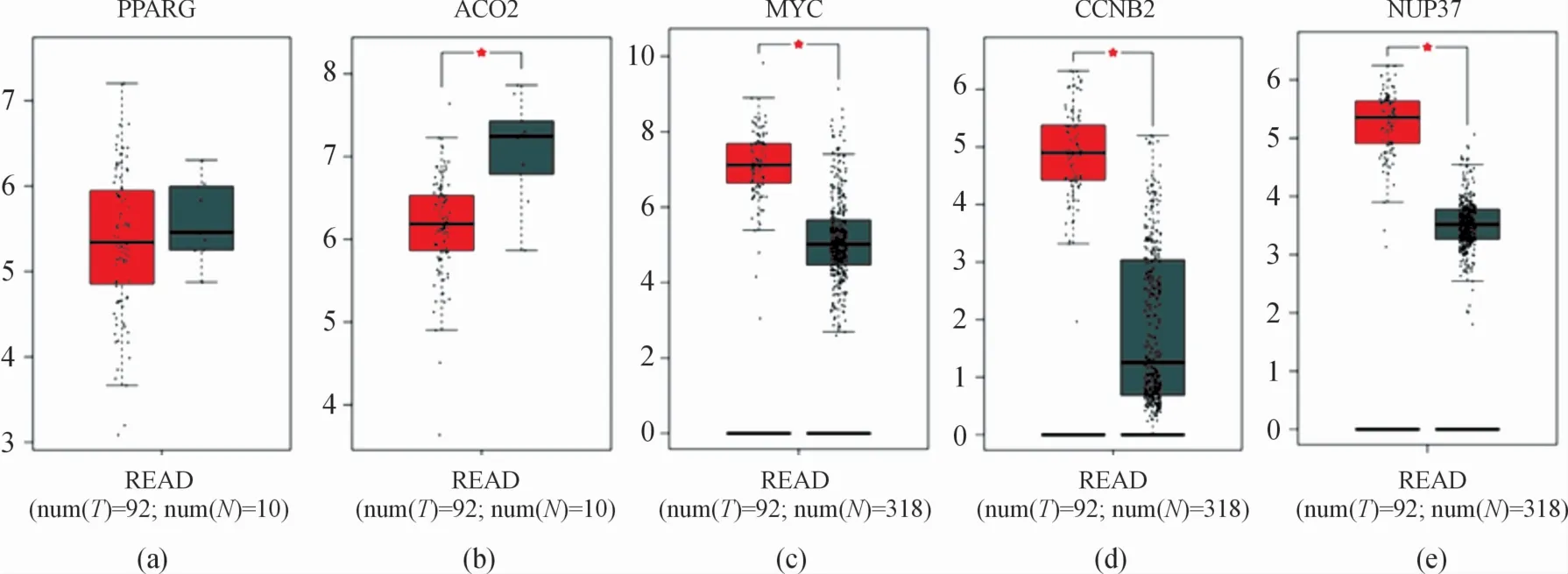
图5 GEPIA数据库验证5个核心基因示意图
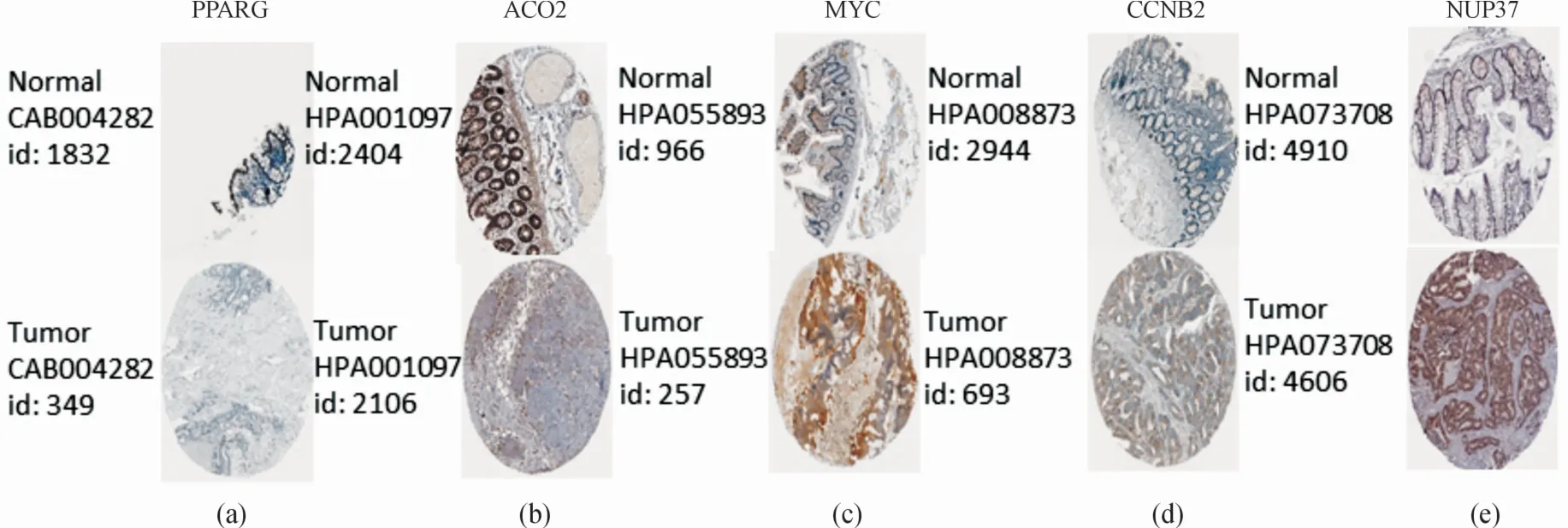
图6 5个核心基因在HPA数据库上的免疫组化染色分析图
3 讨论
结直肠癌是一种发病率较高的消化道恶性肿瘤。由于微阵列被用于预测结直肠癌的潜在治疗靶点,故本研究对GSE32323表达矩阵中方差前25%的基因建立加权共表达网络,以P<0.05选出与结直肠癌显著最相关核心模块进行后续分析,DAVID数据库被用来对核心模块中基因进行功能富集分析,然后用Cytoscape对核心模块中网络节点按照度排序的前30基因分别进行可视化,对所得的60个候选基因在GEPIA数据库通过生存分析,选出Logrank P<0.05的核心基因,最后用GEPIA和HPA数据库和查文献等方式共同验证选出核心基因。
首先,对GSE32323方差前25%的基因进行WGCNA分析得到10个模块,根据P<0.05可得到2个与结直肠癌最相关的核心模块,说明其中的基因与CRC的临床特征具有高度的相关性。功能富集分析结果表明:核心模块中基因主要富集在细胞调控、细胞外泌体、细胞对机械刺激的反应、代谢通路以及富集在细胞分裂、蛋白结合、纺锤、细胞周期和P53信号通路等过程。其中细胞外泌体通路、代谢通路和蛋白结合通路是以前研究没有发现的,而本文中分析找到的这些通路已被证实与CRC有关。Zhi等[9]发现了外泌体miR-25-3p由CRC癌细胞转移至血管内皮细胞中,而且miR-25-3p表达水平明显高于没发生癌症转移的CRC患者。该结论表明外泌体miR-25-3p与癌症转移前微环境形成有关,可能成为一种预测CRC转移的血液生物标志物。Pan等[10]证实肿瘤微环境(TME)诱导的核编码线粒体膜蛋白ANKRD22以自噬独立的方式减少线粒体数量,满足结直肠癌启动细胞(CCICs)的代谢需求,可以促进CRC细胞的代谢重编程。这个结果说明ANKRD22是根除CCICs的潜在靶点。Ma等[11]在体细胞变异体中发现了2种代谢相关基因UQCR5和FDFT1一般只在肝转移中突变,表现出具有临床意义和生物学功能的蛋白丰度失调,在CRC的发生和转移中扮演重要角色。刘会平等[12]发现多配体聚糖结合蛋白(SDCBP)的表达对结直肠癌患者的癌症分期、术后的生存期及淋巴结转移都有显著影响,患者致死的主要缘故为SDCBP的高表达和发生淋巴结转移。该现象表示SDCBP在大肠癌预后中发挥重要功能。王子昕等[13]表明RNA结合基序蛋白6(RBM6)在CRC中呈高表达,并与患者远处转移、临床分期和不良预后相关,可能参与CRC的恶性进展。由此可见,细胞外泌体、代谢通路以及蛋白结合等生物学过程与CRC的发生、发展密切相关。接着用核心模块可视化得到的60个基因在GEPIA数据库中通过生存分析,选出了P<0.05的核心基因分别为PPARG,ACO2,MYC,CCNB2,NUP37。然而,由于CRC类型丰富,临床样本的有限性可能造成CRC的研究不全面,还需实验验证这些核心基因可能对结直肠癌表型和预后具有预测作用。
本研究发现的5个核心基因中有4个已从生物角度被验证与CRC有关。PPARG表观遗传沉默在散发性结直肠癌中起关键作用,并与患者预后显著相关[14]。PPARG作为抑癌基因在结肠肿瘤发生过程中发挥重要作用,并且常表现为表观抑制[15]。研究表明,CRC细胞中FoxO3a的表达在mRNA和蛋白水平上均显著升高,c-Myc是CRC中调节细胞存活、代谢的关键调控因子[16],且FoxO3a与c-Myc相互作用,并在CRC细胞中调控LARS2、MRPL12、PKM2和ACO2等代谢基因。MYC致癌基因是很多癌症发生发展的关键因子,MYC基因过表达情况在常在肿瘤中发现,MYC与肿瘤患者低预后有关[17]。MYC在细胞转化和细胞周期进展、凋亡中发挥作用,许多lncRNAs和转录因子作用于MYC来影响肿瘤生长[18]。Belt等[19]发现III期CRC中高表达的Aurora激酶A(AURKA)与肿瘤复发率增高有关,AURKA有望成为CRC肝转移患者中有效的预后标志物。Takahashi等[20]证实MYC是AURKA/TPX2轴下游的关键分子,抑制AURKA可能成为MYC诱导的肿瘤的靶向治疗新靶点。有报道称细胞周期蛋白B2(CCNB2)在结直肠腺癌中呈显著高表达[21]。辛萱等[22]表明CCNB2的mRNA和蛋白在结直肠癌中呈高表达,其与结直肠癌浸润深度以及肿瘤尺寸有关,可能为结直肠癌的潜在靶点。核孔蛋白基因NUP37虽然之前没有被证实与CRC有关,但它被发现在小鼠模型和肝癌临床标本中显著上调,过表达NUP37促进了肝癌细胞的生长、侵袭和迁移,而抑制NUP37的表达则会提高小鼠模型的存活率。这一现象证实NUP37可能是肝癌的治疗靶点[23]。NUP37存在杂合无意义或剪接位点变异,核膜功能障碍可能是影响遗传性心脏疾病的因素,这一特点表明NUP37是心血管疾病的新候选基因[24]。虽然NUP37在CRC的作用机制还不清楚,但是这一发现为我们探索结直肠癌的发生机制提供了新的思路。
4 结论
本研究通过对GSE32323进行WGCNA分析,功能富集分析和生存分析,得到5个核心基因。其中PPARG、ACO2、MYC和CCNB2在前人研究中从生物验证角度被证实是与CRC预后相关的核心基因,可能作为CRC的生物标志物。目前NUP37在一些疾病中的生物学功能已经被探索,但还缺乏NUP37在CRC发生机制中的功能研究,表明NUP37在CRC诊断、预后和治疗靶点方面值得进一步探索,还需用分子实验确认NUP37在CRC中的功能。在以后的工作中,可对NUP37的功能和机制展开研究,检测其在CRC中的相关通路并对其进行调控,为临床治疗提供有力的理论依据。也可从基因分类的角度把WGCNA方法推广到研究多种癌症处于不同平台的数据,检索其他方法所忽略的重要信息,获取更多的结果。
猜你喜欢
中学生数理化·高一版(2021年2期)2021-03-19
知识经济·中国直销(2018年8期)2018-08-23
中国医药指南(2017年3期)2017-11-13
数学学习与研究(2017年3期)2017-03-09
中国老区建设(2016年1期)2016-02-28
西南军医(2016年3期)2016-01-23
中国继续医学教育(2015年6期)2016-01-07
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年3期)2015-06-10
中国当代医药(2015年30期)2015-03-01
