
Insulin resistance in diabetes: The promise of using induced pluripotent stem cell technology
2021-04-08 01:52AhmedElsayedSelvarajVimalrajManjulaNandakumarEssamAbdelalim
World Journal of Stem Cells 2021年3期
Ahmed K Elsayed, Selvaraj Vimalraj, Manjula Nandakumar, Essam M Abdelalim
Ahmed K Elsayed, Manjula Nandakumar, Essam M Abdelalim, Diabetes Research Center, Qatar Biomedical Research Institute, Hamad Bin Khalifa University, Qatar Foundation, Doha 34110, Qatar
Selvaraj Vimalraj, Centre for Biotechnology, Anna University, Chennai 600025, India
Essam M Abdelalim, College of Health and Life Sciences, Hamad Bin Khalifa University, Qatar Foundation, Doha 34110, Qatar
Abstract Insulin resistance (IR) is associated with several metabolic disorders, including type 2 diabetes (T2D).The development of IR in insulin target tissues involves genetic and acquired factors.Persons at genetic risk for T2D tend to develop IR several years before glucose intolerance.Several rodent models for both IR and T2D are being used to study the disease pathogenesis; however, these models cannot recapitulate all the aspects of this complex disorder as seen in each individual.Human pluripotent stem cells (hPSCs) can overcome the hurdles faced with the classical mouse models for studying IR.Human induced pluripotent stem cells (hiPSCs) can be generated from the somatic cells of the patients without the need to destroy a human embryo.Therefore, patient-specific hiPSCs can generate cells genetically identical to IR individuals, which can help in distinguishing between genetic and acquired defects in insulin sensitivity.Combining the technologies of genome editing and hiPSCs may provide important information about the genetic factors underlying the development of different forms of IR.Further studies are required to fill the gaps in understanding the pathogenesis of IR and diabetes.In this review, we summarize the factors involved in the development of IR in the insulin-target tissues leading to diabetes.Also, we highlight the use of hPSCs to understand the mechanisms underlying the development of IR.
Key Words: Type 2 diabetes; Insulin target tissues; Human pluripotent stem cells; Induced pluripotent stem cells; Genetic factors; Disease modeling
INTRODUCTION
Insulin resistance (IR) is a hallmark of type 2 diabetes (T2D) and other related metabolic disorders.Several hereditary and environmental factors are known to be involved in the development of IR in individuals at risk for T2D.Persons at genetic risk for T2D tend to develop IR several years before glucose intolerance[1].Diabetes is associated with several complications, such as diabetic ketoacidosis, nonketotic hyperosmolar coma or death, heart disease, stroke, kidney failure, foot ulcers,etc.Glucose metabolism is regulated by a feedback loop between islet β-cell and insulintarget tissues.Under IR condition, the β-cells control normal glucose tolerance by increasing the level of insulin secretion[2].IR in different tissues (adipose tissue, skeletal muscle, liver, brain, gut, pancreas, vasculature, and kidney) leads to several metabolic disorders, including T2D, cardiovascular diseases, hypertension, polycystic ovary syndrome (PCOS), fatty infiltration of the liver, non-alcoholic fatty liver disease (NAFLD), apnea, a sleep disorder, arthritis, skin diseases, and cancers.Previous studies showed alterations in the gene expression profile between individuals with a family history of T2D and those without a family history of the disease.Those defects were mainly observed in the genes related to mitochondrial function and fat metabolism[3].However, it is difficult to distinguish whether the alterations in the gene profiles are due to genetic or environmental factors.Although several genetic and environmental factors are known to be involved in the development of IR, the molecular and cellular mechanisms underlying IR development and its progression to T2D remain not completely understood.This is due to the lack of appropriate human models to study the pathophysiology of different forms of IR.
The establishment of induced pluripotent stem cell (iPSC) technology has allowed the generation of pluripotent stem cells (PSCs) from somatic cells and has led to the establishment ofin vitromodels to study the genetic factors involved in the development of human diseases[4].The fact that iPSCs can be produced without the need of a human embryo enables us to avoid ethical concerns that restricted researchers, for decades, to use human embryonic stem cells (hESCs) in stem cell research studies.iPSCs have unlimited proliferative ability and a great potential to differentiate into all cell types of the body[4].Therefore, iPSCs provide a source of a human model to study the IR in insulin target tissues and pancreatic β-cell dysfunction.iPSCs can generate cells genetically identical to insulin-resistant individuals, which can help in distinguishing between genetic and acquired defects in insulin sensitivity.In the current review, we mainly focus on the IR associated with diabetes and the mechanism involved.Also, we discuss the use of iPSC technology to understand and treat these disorders and explain the challenges and limitations of using the human iPSC-based models.
INSULIN RESISTANCE IN THE INSULIN-TARGET TISSUES
Insulin, secreted from pancreatic β-cells, plays a critical role in a wide range of cells and tissues in the body through its main action in regulating the cellular energy and metabolic processes of the macronutrients (carbohydrate, protein, and lipid) besides its growth-promoting action through its mitogenic effect[5].Impairment in insulin secretion and/or action can influence the functionality of most organs and affects the normal physiology of the whole body.The insulin performs its functions in insulintarget tissues through its binding to the insulin receptor (INSR), which activates its downstream signaling pathways (Figure 1).Insulin has an anabolic effect as it promotes glycogen, lipid, and protein synthesis as well as inhibits gluconeogenesis and lipolysis processes[6].It also facilitates the intracellular glucose uptake through translocation of the insulin-dependent glucose transporter (GLUT4) as in skeletal muscle and adipose tissue[7].This metabolic action of insulin is achieved through the phosphatidylinositol 3 (PI3K) kinase pathway[8].The growth-promoting action of insulin is achieved through INSR and insulin growth factor receptor, leading to activation of Ras/MAP kinase pathways and subsequently activates the transcription factors of cell division and proliferation[9].
IR is the condition in which the cells respond inappropriately to the circulating insulin.In other terms, it is the impaired sensitivity to insulin-mediated actions[10].It is known that insulin controls energy production mainly through glucose oxidation and inhibiting other sources such as lipolysis, protein catabolism, glycogenolysis, and gluconeogenesis.However, under IR conditions, these processes are activated as an alternative source of glucose.These catabolic processes are accompanied by the accumulation of toxic metabolic byproducts and inflammatory factors, which have harmful effects on the insulin-target tissues.In skeletal muscles, the muscular glycogen and protein synthesis are impaired with a decrease in the glucose uptake leading to sarcopenia[9].Under IR conditions, the lipolysis is enhanced leading to the release of triglyceride, free fatty acids (FFAs), and inflammatory cytokines (e.g.,IL-6, TNFα, Leptin) into the circulation[11].The metabolic toxic derivatives of FFAs and the inflammatory cytokines influence the functionality of most of the tissues either directly through its lipo-toxic and lipo-apoptotic effects or indirectly through an impairment in the insulin signaling pathway[12,13].The liver responds to IR and the demand of other cells to glucose by stimulation of the glycogenolysis process to produce more glucose[14].Under IR conditions, the released FFAs from fat are transported to the liver and cause NAFLD (steatohepatitis), which is subsequently followed by liver cirrhosis[15].This impairment in liver function leads to a decrease in insulin clearance with hyperinsulinemia.Furthermore, IR induces impaired mitochondrial oxidative metabolism and endocrine disorders like PCOS, adrenal disorders, and thyroid function abnormalities[16].It has been reported that the insulin-sensitive brain regions include the hypothalamus, prefrontal cortex, hippocampus, and fusiform gyrus.Therefore, the IR in the brain leads to mild cognitive impairment and dementia, and Alzheimer's disease[17].IR in the gut leads to alteration in microbiota resulting in dysregulation in the short-chain fatty acid production and gut hormone production[12,18].The clinical disorders associated with IR include T2DM, cardiovascular diseases, hypertension, PCOS, fatty infiltration of the liver NAFLD, apnea, a sleep disorder, arthritis, skin diseases, and cancers.
β-CELL DYSFUNCTION ASSOCIATED WITH INSULIN RESISTANCE
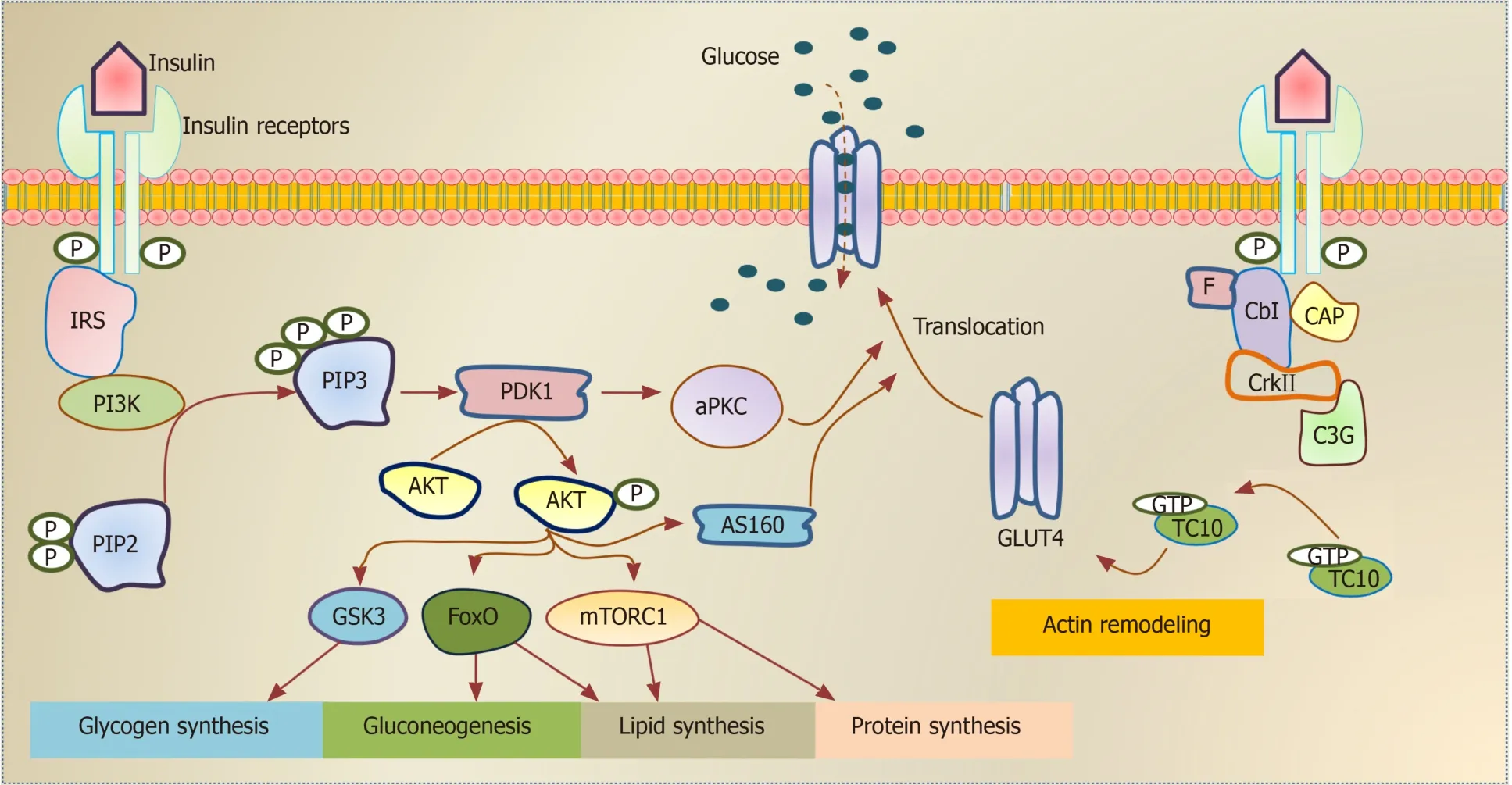
Figure 1 Schematic illustration of insulin signaling pathways.
Impairment in glucose homeostasis regulated by the feedback loop between the insulin target tissues and pancreatic β-cells leads to T2D, which is associated with an abnormal increase in blood glucose levels (Figure 2).IR and pancreatic β-cell dysfunction are the major characteristic features of T2D pathological conditions.In T2D, β-cell dysfunction occurs as a result of IR in the insulin-target tissues[19].However, the interplay between IR and pancreatic β-cell dysfunction is still complex (Figure 2).Many metabolic insults, such as obesity, saturated FFA overconsumption, inflammatory cytokines, and oxidative stress and endoplasmic reticulum (ER) stress reduce the functionality of β-cells and dysregulate the normal physiological state of βcells, leading to their demise[20].Loss of insulin sensitivity or IR results in hyperglycemia, hyperinsulinemia as well as activation of the catabolic processes, which increase the level of FFAs and lipotoxic cytokines.All of these elevated elements caused by IR are responsible factors for β-cell stress and dysfunction[21].In the early stages of IR, β-cells try to compensate and control glucose homeostasis through the production of more insulin[22].However, with chronic prolonged exposure to hyperglycemia, β-cells secrete large quantities of insulin, leading to ER stress and exhaustion of β-cells with depletion of insulin store[23].Moreover, hyperglycemia for a long time leads to a decrease in the activity of insulin promoter through the reduction in the binding of PDX1 and MAFA with a subsequent decrease in insulin gene expression and its secretion[24].Hyperglycemia also can induce oxidative stress, inflammation, pro-apoptotic, and apoptotic genes’ expression in β-cells[25].Elevated plasma level of FFAs and glucose as a result of IR lead to glucolipotoxicity leading to β-cell failure[26].These saturated FFAs induce both ER and oxidative stress in human βcells and islets through the overproduction of NOS2 and NO in β-cell mitochondria[23,27]or through compromising the ER morphology and integrity[27].In the presence of hyperglycemia, FFAs influence the biosynthesis and expression of the insulin gene, leading to suppression of adequate insulin secretion in response to glucose[28].Increased FFAs lead to intrapancreatic and intra β-cell accumulation of triglyceride and fat droplets, triggering β-cell dysfunction and death due to an increase in the inflammation process[24,29].Inflammation and the proinflammatory cytokines are recognized as an important contributor to β-cell dysfunction[30].IR-associated inflammatory cytokines, such as IL6, TNFα, IFNγ, NF-κB, and others cause the dysfunctionality and death of β-cellsviadamage in the mitochondria, cellular proteins, lipids, nucleic acids, and ER stress[31].The inflammatory cytokines and the recruited immune cells in the inflamed dysregulated pancreatic islet trigger β-cell dysfunction[32].Proinflammatory cytokines mediate reactive oxygen species and reactive nitrogen species production and reduce ATP production and eventually lead to β-cell dysfunction[33].The β-cell dysfunction and inadequate β-cell mass expansion can be due to the defect in the insulin signaling pathway in the pancreatic β-cells.Improper glucose sensing has been noticed in mouse β-cells, which lack the INSR or IGF1R.Additionally, loss of INSR leads to the β-cell mass reduction and early onset diabetes[34,35].Another study showed similar diabetic phenotypes in mice with PDK1 deficiency in β-cells[36].The impairment of cell cycle progression is hypothesized to be engaged in the β-cell mass reduction and dysfunction.It has been found that the cell cycle inhibitor, p27Kip1, is accumulated in the nucleus of the β-cells of hyperglycemic IRS2-deficient mice and the deletion of the p27Kip1 gene ameliorates β-cell proliferation and the hyperglycemia, reflecting the role of cell cycle inhibition in the βcell function[22].
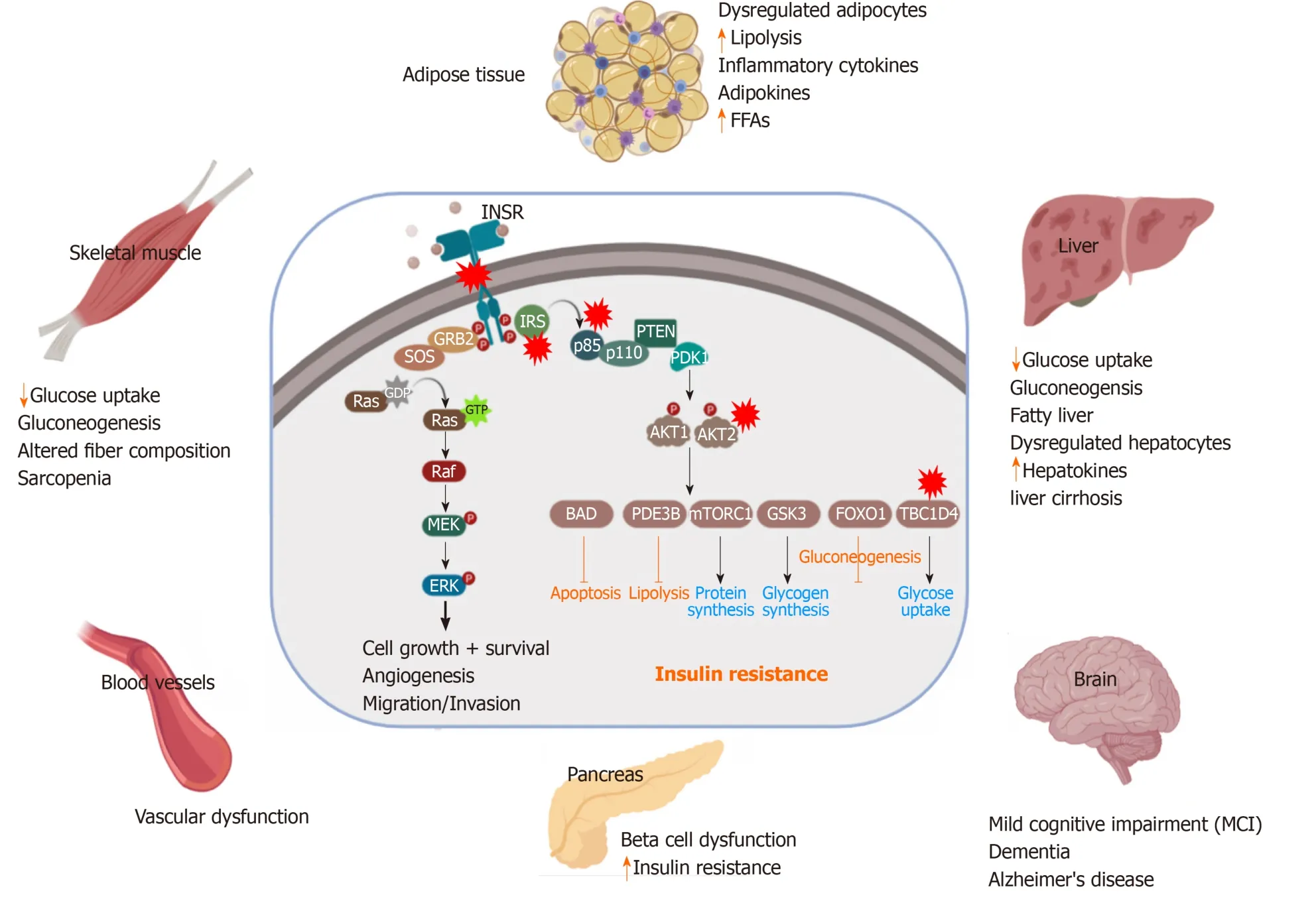
Figure 2 Pathological effect and consequence of insulin resistance.
STUDYING THE MECHANISMS UNDERLYING THE DEVELOPMENT OF INSULIN RESISTANCE
The IR arises as a result of the inability of the insulin-target tissues to use insulin appropriately, which is caused by many factors.The causes of IR involve genetic and environmental factors.The environmental factors can influence the insulin signaling pathway such as unhealthy diets, obesity, lack of physical activity, pharmacological agents, stress, cytokines, and hormones.These environmental factors combined with genetic factors can lead to the development of IR and diabetes[37].Genetic predisposition causing IR may have a direct effect on the insulin signaling pathway or indirectly affecting other targets with a subsequent secondary effect on the insulin pathway[38].The disruption and/or mutations in the genes encoding proteins involved in insulin signal transduction impair insulin action and reduce the rate of glucose uptake leading to IR[38].Previous studies used multiple approaches to investigate the molecular mechanisms underlying the development of different forms of IR.These approaches include animal models, human insulin target tissues’ explants, the human population genetic studies, and stem cells.
Animal models of insulin resistance and their limitations
Most of the animal models used to study IR are derived by inducing stress, diet, injuries, chemicals, and by different combinations mimicking IR conditions[39].Meanwhile, there are few genetically modified IR animal models such asIRSknockout mouse (IRS-/-)model Akt2 knockout (Akt2-/-) mouse model,Glut4+/-null mutant mouse, andGlut2-/-mice[39,40].Lipodystrophy has shown a strong association with severe IR; therefore, transgenic animals with defects in the genes regulating fatty acid metabolism are used to study IR mechanisms, such as sterol regulatory-element binding protein 1, A-ZIP/F-1, and toll-like receptor 4[39].There are several animal models exhibiting the features of the IR and diabetes due to mutations in specific genes either spontaneously or through selective breeding such as Leptin ob/ob and leptin db/db mouse strains, Zucker fatty (fa/fa) rat, New Zealand mice strain, OLETF rat, KK/Ay mouse and other models of the spontaneously developed IR strains[39].Although the above-mentioned animal models provided a lot of information about the mechanisms of IR, they could not resolve all the questions related to the development of IR in human tissues and the relationship between IR and β-cell dysfunction.
Studying insulin resistance in human insulin-target tissues
The molecular and genetic basis for the development of IR in the insulin-target tissues such as skeletal muscle, adipose tissue, and liver are not fully understood.Previous studies used muscle biopsies showed that the mitochondrial DNA, mitochondrial genes, and respiratory chain subunit proteins are downregulated in IR subjects compared to healthy controls[41].Another study showed that cultured myoblasts isolated from skeletal muscle of T2D patients have defects in glucose transport and insulin signaling[42].In the skeletal muscle of the IR offspring of T2D parents, it has been found that the mitochondrial ATP production, mitochondrial density, and AKT activation are significantly reduced, while the IRS-1 phosphorylation (serine) is significantly increased[41,43].The activation of PI3K pathway is required for the transport of glucose and synthesis of glycogen, which is impaired after insulin stimulation in insulin-resistant skeletal muscle[44].The upregulation of IRS-1 phosphorylation on serine residues has been found to suppress insulin signaling in T2D[43].Taken together, these findings indicate that the defects in insulin signaling, glucose transport, glycogen synthesis, and mitochondrial activity are the main dysfunctional features associated with the IR in the skeletal muscle.
Adipose tissue is considered the main regulator of insulin action in the body; therefore, defects in insulin signaling in adipocytes cause systemic IR, indicating that impairments in adipose insulin signaling are a common hallmark of IR[45].Glucose transporter, GLUT4, is significantly downregulated in subcutaneous adipose cells from T2D patients and in healthy individuals with a genetic susceptibility for T2D[46].This reduction in GLUT4 expression is associated with changes in the secretion of adipokines.Also, activation of peroxisome proliferator activated receptors (PPARs), which are responsible for adipogenesis regulation as well as lipid metabolism in adipocytes, leads to an improvement in insulin sensitivity through the induction of the expression of several genes that are related to the insulin signaling pathway.In addition to its role in adipogenesis, PPAR is also involved in regulating lipid metabolism in mature adipocytes by increasing fatty acid trapping[47].The importance of insulin signaling in the development of adipocyte IR has been reported in T2D, where previous studies reported that the substrate of IRS-1 is involved in IR in adipocytes by inhibiting insulin-signaling[48].Also, in T2D patients, several IRS-1 mutations have been found[49]and a reduced IRS-1 protein level has been observed in adipocytes from other T2D patients[50], relatives of T2D patients, and obese individuals[51].In addition to insulin signaling defects, dysregulation of adipokines and the lipolysis process are linked to IR and T2D.The adiponectin gene is considered a candidate susceptibility gene for T2D as it has been detected on chromosome 3q27, which is linked to T2D and metabolic disorders[52,53].
The insulin signaling in the hepatic cells is crucial for maintaining normal liver function and regulating glucose homeostasis[54].The liver activates glucose uptake and glycogen storage, but it inhibits glycogenolysis and gluconeogenesis.Hepatic IR progression plays a critical role in the pathogenesis of T2D.Loss of INSR signaling in hepatic tissue leads to an increase in gluconeogenesis and a decrease in lipogenesis[54,55].The IRS-1/2 is involved in the suppression of gluconeogenesis and stimulation of lipogenesis.In the liver, insulin inhibits IRS-2 expression at the transcriptional level and doesn’t influence IRS-1 expression[55,56].Hepatic IR is characterized by the inability of insulin to inhibit the production of glucose in the liver[57].This impairment in liver function leads to a decrease in insulin clearance with hyperinsulinemia.The released FFAs from fat tissues due to IR transports to the liver cause NAFLD (steatohepatitis), which is followed by liver cirrhosis[15].Liver-derived proteins termed hepatokines are released into the circulation, which cause defective insulin signaling[58].
USING INDUCED PLURIPOTENT STEM CELLS (iPSCs) TO STUDY INSULIN RESISTANCE
Although animal models provided knowledge on the pathogenesis of certain forms of IR and diabetes, they cannot reflect all pathophysiological features of human diseases due to the physiological differences between humans and animals.It has been reported that the genetic makeup of the mouse model is involved in the alterations of the phenotypical outcome, even when the same mutation is created in mice with different genetic makeup.Several attempts have been made to overcome these challenges by the generation of humanized mouse models[59]or by using non-human primates[60].Several population-based studies have been reported, in which the biopsies are isolated from both insulin-resistant and insulin-sensitive individuals to determine the mechanisms underlying IR[61].The primary endothelial cell-based methodologies have been used to study the vascular inflammation in diabetes and atherosclerosis[30].However, it also has certain limitations such as lack of donor availability and limited lifespan of primary cells.Furthermore, it is difficult to access human tissues, such as hepatocytes and skeletal muscles, particularly at the preclinical stages of the disease.In addition, it is difficult to distinguish between genetic and environmental factors.
In order to overcome the above-mentioned limitations, establishing a human cell model offers a great opportunity to understand the pathophysiology of human diseases.The hiPSC technology can provide human cell models to study the pathophysiology of IR.Generating patient-specific iPSCs from IR individuals with specific mutations or a family history of IR and diabetes can be used to study the genetic factors involved in the development of the disease, because the iPSCs maintain all the genetic information of the patients.Those patient-specific-iPSCs can generate all insulin target cells, such as skeletal myotubes, adipocytes, and hepatocytes as well as other cell types.The generated cell types carry the same genetic signature of the patients allowing further studies to identify the genetic defects involved in the disease progression (Figure 3).
iPSCs have been successfully generated from patients with different forms of diabetes including monogenic forms of diabetes, mitochondrial diabetes[62,63]T2D patients[64]as well as T1D patients[65].The monogenic forms of IR can be modeled using the iPSC technology (Table 1).The IR is associated with several monogenic disorders such as Donohue and Rabson Mendenhall syndromes (INSR mutation), SHORT syndrome (PI3K mutation), Alstrom syndrome (ALMS1 mutation), Werner syndrome and Bloom syndrome (defects in DNA helicase), congenital generalized lipodystrophy (CGL) (AGPAT2 or BSCl2 mutations), and familial partial lipodystrophies (FPLD) (LMNA and PPARG mutations)[38].In addition, several mutations associated with severe IR have been reported in AKT2, AS160 (TBC1D4), PPARG, PPP1R3A, and POLD1 encoding DNA polymerase delta[38].Recent studies showed the generation of iPSCs from patients with IR; however, all of those studies focused on a very specific form of IR, which is due to specific mutations in INSR[66-69](Table 1).Two of those studies generated iPSCs from patients with INSR mutations (INSR-Mut) (Donohue syndrome) focused on the effect of INSR-Mut on pluripotency and mitochondrial function in undifferentiated INSR-Mut hiPSCs[68,69].It has been shown that INSR-Mut iPSCs are defective in their self-renewal ability because insulin and its downstream signaling are involved in regulating the unique properties of self-renewal and pluripotency in the undifferentiated iPSCs[70].The PI3K has been shown to be crucial for the self-renewal of pluripotent stem cells[70].Also, the INSR-Mut-iPSCs have mitochondrial dysfunctions, including alterations in the number and the size of mitochondria, and were associated with an upregulation in the expression of mitochondrial fission factor and inverted formin 2[68].These two genes are known to enhance the mitochondrial fission as indicated in the INSR-Mut-hiPSCs[68].Interestingly, increased mitochondrial fission has previously been detected in adult tissues, such as pancreatic β-cells and skeletal muscles of T2D patients[71].Furthermore, it has been reported that mitochondrial DNA variation could be associated with genetic alteration, a known risk factor for T2D[72].Also, the expression of glycolytic enzymes is downregulated, while lactate production is increased.These events lead to enhancement of ADP/ATP ratio and 5' AMP-activated protein kinase activity as well as leading to inefficient ATP and decrease in energy production with an increase in the oxidative stress[67].All the derived INSR-Mut hiPSCs showed reduced proliferation and defective INSR phosphorylation and defects in its downstream signaling pathway such as AKT, GSK3, ERK1, and ERK2[69].Differentiation of INSR-Mut hiPSCs towards skeletal myotubes exhibits defects in insulin signaling, glucose uptake, glycogenaccumulation, and altered insulin signaling gene expression[66], indicating the genetic defects in the skeletal myotubes.Another iPSC model for IR, in which iPSCs have been generated from the fibroblasts of an insulin-resistant patient with CGL, an autosomal recessive disease due toBSCL2mutation[73].These patient-specific iPSCs have been used as anin vitromodel to study the physiopathology of lipid accumulation and lipodystrophy and its relation to IR.Adipocytes derived from these BSCL2-Mut iPSCs showed reduced lipid droplet formation and dispersed cytoplasmic distribution of adipose differentiation-related protein[74].Another study generated iPSCs from a patient with familial FPLD2 due to mutation in theLMNAgene[75].These FPLD2-iPSCs recapitulated the insulin resistance phenotype of the patient with low efficiency ofin vitroadipogenic differentiation and less functionality[75].Recent studies showed the ability to generate adipocytes from hESCs and hiPSCs[76,77].It has been reported that hiPSC-derived adipocytes, transplanted into mice, are able to sustain their functional characteristics for several weeks[77], suggesting that these cells can also be used therapeutically to improve metabolic disorders in patients.Therefore, differentiation of patient-specific hiPSCs into white adipocytes can offer a large number of functional adipocytes for transplantation as a possible way to treat adipocytes-associated disorders as well as studying IR.Since the liver is an important insulin target tissue, the iPSCs have been used to understand the etiology of steatosis due to the NAFLD, which is accompanied by IR and hyperlipidemia[78].The iPSCs generated from patients with liver steatosis showed a decrease in the AKT/mTOR signaling pathway and the IR phenotypes are observed in both liver and skin fibroblasts of the patients.Additionally, it has been shown that the transcription factor, sterol regulatory elementbinding transcription factor 1, and its downstream targets, LIPIN1 (LPIN) and low
density lipoprotein receptor, are involved in glycerolipid and fatty acid biosynthesis[78].
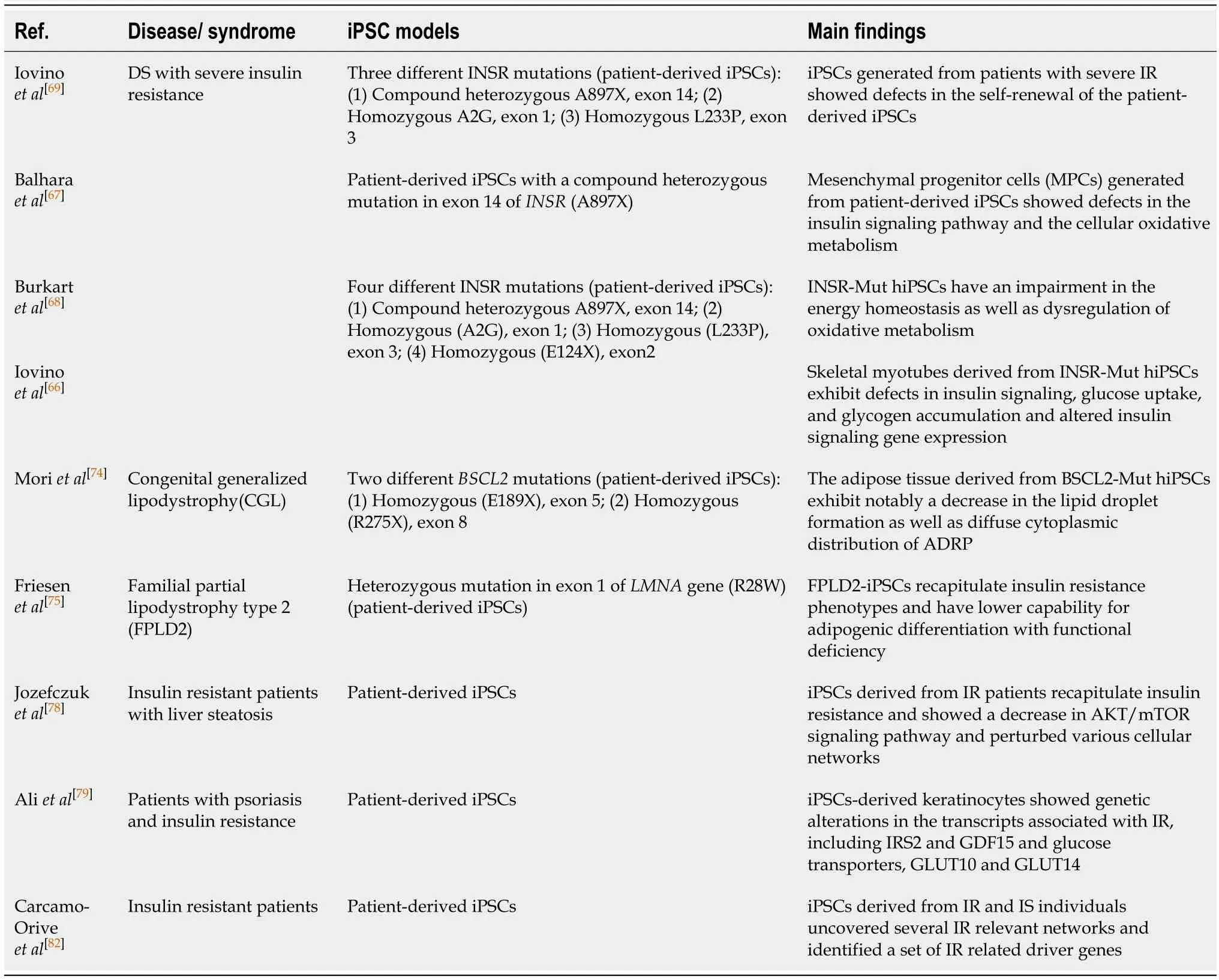
Table 1 Human induced pluripotent stem cells models used to study insulin resistance
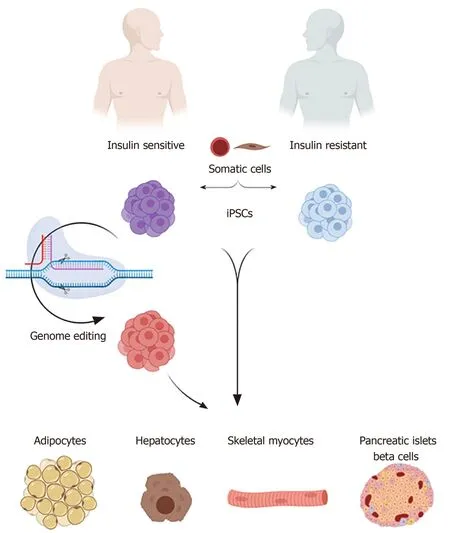
Figure 3 Using human induced pluripotent stem cells to study insulin resistance.
iPSCs have been also utilized to understand the polygenic form of IR.In our recent study, we used iPSC technology to understand the pathogenesis of psoriasis, skin disease, and its link to IR[79].The results showed that keratinocytes derived from patient-specific iPSCs with psoriasis have genetic alterations in the transcripts associated with IR, includingIRS2andGDF15and glucose transporters,GLUT10andGLUT14[79].These findings indicate that patient-specific iPSCs can provide important information on the genetic predisposition of IR.
Further studies are required to fill the gaps in understanding the pathogenesis of IR and diabetes.There are several genetic factors involved in the development of IR; therefore, patient-specific iPSCs can be used to study the most common form of IR leading to T2D.Also, hiPSCs/hESCs can be genetically edited using genome editing tools, such as CRISPR/Cas9[80].The recent genome-wide association studies (GWAS) studies have discovered several genetic defects associated with IR and diabetes development.Those defective genes can be introduced into hiPSCs/hESCs using a genome-editing tool followed by their differentiation into insulin-target cells, such as skeletal muscles, adipocytes, and hepatocytes.The combination of these technologies may provide more details about the inherited factors underlying the development of different forms of IR with a particular focus on the common form of IR.The generation of isogenic hiPSCs represents a proper human cell model to study the molecular mechanism of the newly identified candidate genes and variants through GWAS[81].A recent study showed the generation of a large number of iPSC lines from individuals with and without IR[82].Comparing the transcriptome profiles between iPSCs derived from 52 IR and 48 insulin sensitive individuals showed 1388 differentially expressed genes between both groups[82].Nine of those genes (BNIP3, CARS, IDH1, NDUFB1, HMGCR, HPN, FDPS, SLC27A1,andTMEM54) have been shown to be associated with IR and T2D[82].These hiPSC lines are very useful to investigate the underlying mechanisms of IR and its associated metabolic disorders.
LIMITATIONS OF IPSCS AS AN IN VITRO MODEL TO STUDY INSULIN RESISTANCE
There are some challenges of using iPSC-based models.For example, robust protocols are needed for the differentiation of iPSCs into the desired cell types for studying metabolic diseases.Currently, the protocols are available for differentiation of hPSCs into the main insulin-target cells (adipocytes, skeletal muscles, and hepatocytes)[76,83-85]as well as pancreatic beta cells.However, most of those protocols lead to a combination of diverse cell types.The variability in the biological properties of iPSCs derived from different individuals shows the difference in the differentiation nature towards a given lineage.This reasonably affects the consistency of interpretation of the phenotypes[86].The results obtained from populations of different genetic backgrounds may show differences.For instance, a report emphasized that population-based gene sequencing showed significant variations in the human genome[87].Hence, generating iPSCs from patients with genetic defects, differentiating them to specific cell types, may hold a noteworthy risk[88].Generation of an iPSC model for IR is a comparatively difficult process, because the development of metabolic diseases typically develops overtime and may show weak phenotypic changes underin vitroconditions[88].Genome editing in iPSCs can be used to study candidate genes involved in IR and diabetes[87].Also, the generation of isogenic cell lines from iPSCs could be an important strategy to overcome the problem of differences in the genetic background between different iPSC lines.The isogenic cell lines possess similar genetic milieu, epigenetic nature, and differentiation properties.This strategy may provide more consistent output and interpretation for complex diseases.The genome editing tools can make this strategy possible by engineering the genome of iPSCs.There are several reports highlight the use of genome editing tools for the generation of iPSC-based disease models[88,89].
CONCLUSION
Metabolic diseases like diabetes lead to several life-threatening complications.To develop treatments for those metabolic disorders, it is important to understand the factors involved in the development of the disease.IR is associated with several metabolic disorders; however, the genetic factors contributing to IR development are largely unknown.The human iPSC technology can provide cells genetically identical to patients with IR and diabetes.Those patient-specific iPSCs can be differentiated into all cell types involved in the IR, such as skeletal muscle, adipocytes, and hepatocytes as well as other cell types.The IR developed due to a mutation or a defect in one gene (monogenic) can be studied using iPSCs.However, the common form of IR, which is associated with genetic defects in several genes is more difficult to be studied using the iPSC technology.The recent advances in genome editing tools allow us to introduce or correct mutations in patient-specific iPSCs and hESCs as well as to generate knockout hiPSC models.Moreover, recent GWAS studies have identified several new genes involved in the development of IR and diabetes.Therefore, combining hiPSC technology, genome editing tools, and GWAS studies can help in understanding the genetic defects associated with IR.However, the common form of IR is not caused by a single gene defect, but a group of genes is involved in IR progression.Furthermore, establishing efficient differentiation protocols for the insulin-target cells is important to study the genetic defects.Currently, most of the differentiation protocols for those cells are well-established; however, the heterogeneity of the generated cells still need to be resolved.One of the solutions is to purify the target cells using surface markers or other methods.In conclusion, although there is progress in the use of hiPSC technology to study IR, a lot of work still needs to be done.
