
白茅根HPLC指纹图谱及炮制前后化学成分分析
2021-04-08 03:02:24罗良渊徐文芬陈胤睿郑亮亮韦华兰
亚太传统医药 2021年3期
罗良渊,刘 趣,徐文芬,陈胤睿,郑亮亮,杨 港,韦华兰
(贵州中医药大学 药学院,贵州 贵阳 550025)
白茅根为常用中药,又名兰根、地筋、茅草根、甜草根等,来源于禾本科植物白茅ImperatacylindricaBeauv.var.major(Nees) C.E.Hubb.的干燥根茎,为2015年版《中国药典》一部收载品种。白茅根饮片有白茅根和茅根炭两种规格,具有凉血止血、清热利尿的功效,临床用于治疗血热吐血、衄血、尿血、热病烦渴、湿热黄疸、水肿尿少、热淋涩痛[1]。早在《神农本草经》中就有记载茅根味甘,性寒,主劳伤虚羸,补中益气,除瘀血,血闭寒热,利小便[2],其用药历史悠久,疗效可靠。白茅根亦可鲜用,与玉米须同食,取其清热利尿之效。
白茅根的现代研究主要集中在化学成分、药理作用、质量控制等方面。白茅根化学成分主要有绿原酸、棕榈酸、反式对羟基桂皮酸等有机酸类[3-5],多糖、葡萄糖、果糖和木糖等糖类[6]。此外,该药材中还含有三萜类、甾醇类等[7-9]化合物。现代药理学研究表明白茅根具有止血、利尿降压、抗炎镇痛、抗肿瘤、降血糖降血脂等[10-15]药理作用,临床用于治疗肝炎、急性肾炎和血尿等疾病[16-19]。在质量控制方面,2015年版《中国药典》一部收载的白茅根质量标准中包括来源、性状、显微鉴别、薄层鉴别、水分、灰分和浸出物含量的测定,这对于成分复杂的中药来说难以全面反映其真实质量。关于白茅根质量控制方面的文献报道有单个及总类成分含量测定、指纹图谱等,尤以含量测定的报道居多,如采用高效液相色谱法测定白茅根中联苯双酯、4,7-二甲氧基-5-甲基-香豆素等含量[20-21];采用紫外分光光度法测定白茅根中总酚酸、多糖的含量等[22-23];张素红、赵燕燕等[23-24]采用HPLC法进行了白茅根药材、氯仿提取物指纹图谱研究;曹雨诞等[25]采用HPLC法研究了白茅根炒炭前后5-羟甲基糠醛的含量变化。这些研究取得的进展在一定程度上为白茅根药材质量评价提供了参考,但是对于白茅根鲜药材及其炮制前后整体化学成分变化的研究鲜有报道。鉴于此,本文采用高效液相色谱法建立鲜白茅根、白茅根和茅根炭的化学指纹图谱测定方法,构建其不同炮制品的指纹图谱,并对其指纹图谱进行对比分析研究,旨在为白茅根药材及饮片的质量控制及评价提供理论依据。
1 仪器与材料
1.1 仪器
Thermo Ultimate-3000型高效液相色谱仪(美国赛默飞公司);AG1353型电子分析天平(瑞士Mettler-Toledo公司);DK-98Ⅱ水浴锅(天津市泰斯特仪器有限公司);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 药材
实验样品由课题组采自贵州主要分布区及山东购买,经贵州中医药大学孙庆文教授鉴定为禾本科植物白茅ImperatacylindricaBeauv.var.major(Nees) C.E.Hubb.的干燥根茎,除去须根和膜质叶鞘,采取相应方法处理,备用。药材来源信息见表1。
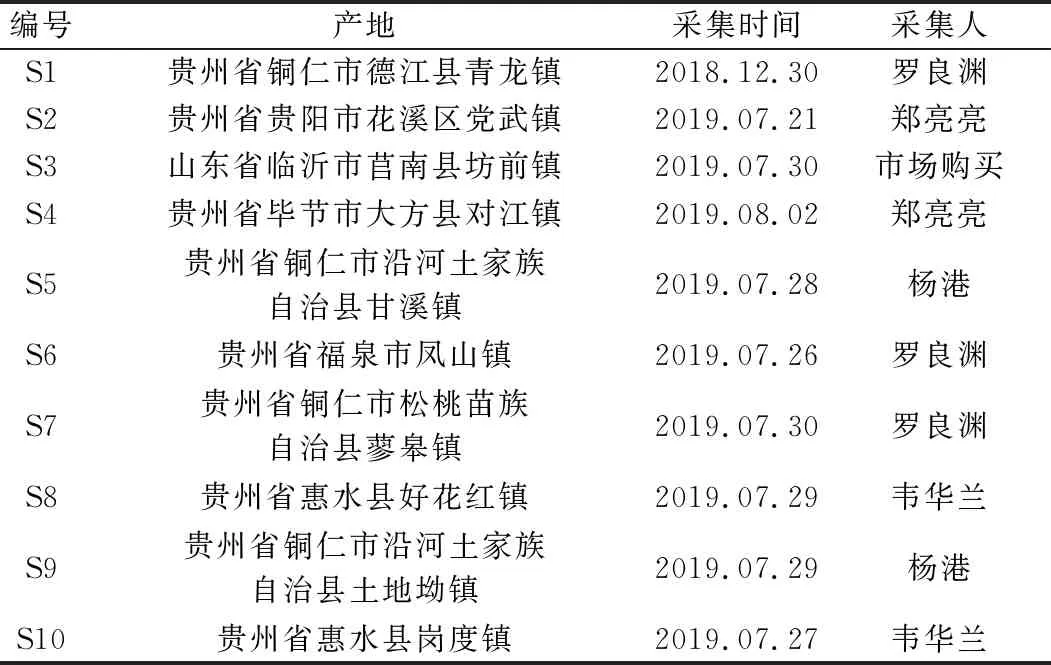
表1 样品信息
1.3 试剂
绿原酸对照品(HPLC≥99.39%,批号MUST-18030620);五羟甲基糠醛对照品(HPLC≥98%,批号BGY-000589);乙腈、甲醇为色谱纯(美国天地有限公司);磷酸、甲醇、乙醇等均为分析纯,水为重蒸馏水。
1.4 样品处理
1.4.1 鲜白茅根 将采集的白茅根茎洗净,除去须根和膜质叶鞘,微润切段,放入冰箱冷藏室备用。
1.4.2 白茅根 将采集的白茅根茎洗净,除去须根和膜质叶鞘,微润切段,称重并记录。置于55 ℃烘箱中干燥,水分不超过12.0%,称重并记录,计算各批次茅根干燥减失的水分含量(干燥减失的水分含量=(鲜白茅根质量-干燥后白茅根质量)/鲜白茅根质量×100%),粉碎过三号筛备用。
1.4.3 茅根炭 取“1.4.2”项下白茅根适量,参照2015年版《中国药典》四部炒炭法(炮制通则0213),中火炒至表面焦褐色,内部焦黄色,放凉,粉碎过三号筛备用。
2 方法与结果
2.1 供试品溶液制备
2.1.1 白茅根 称取1.0 g白茅根药材粉末,精密称定,精密量取20 mL 70%乙醇,水浴回流3 h,恢复至室温,用相应溶剂补足减失的重量,过滤,滤液至水浴锅上挥干,用70%乙醇定容至5 mL容量瓶中,0.45 μm微孔滤膜滤过即得供试品溶液。
2.1.2 鲜白茅根 根据相应批次茅根的干燥减失的水分含量,计算出1.0 g干品所对应的鲜品质量,称量,用研钵研碎,转移至50 mL的锥形瓶中,按照“2.1”项下方法制备供试品溶液。
2.1.3 茅根炭 按照“2.1.1”项下方法制备。
2.2 对照品溶液制备
精密称取绿原酸对照品适量,加70%乙醇使溶解并得浓度为20.0 μg·mL-1的绿原酸对照品溶液,备用。
精密称取5-羟甲基糠醛对照品适量,加70%乙醇使溶解并得浓度为60.0 μg·mL-1的5-羟甲基糠醛对照品溶液,备用。
2.3 HPLC色谱条件
色谱柱:Diamonsil C18(250 mm×4.6 mm,5 μm),流动相:乙腈-0.05%磷酸水溶液,流速:1.0 mL·min-1,柱温:25 ℃,检测波长:280 nm,进样量:10 μL。梯度洗脱程序见表2。

表2 梯度洗脱程序
2.4 方法学考察试验
2.4.1 精密度试验 取S5白茅根样品1份,按照“2.1”项下方法制备供试品溶液,按照“2.3”项下色谱条件连续进样6次,采集并记录色谱图,并导入《中药色谱指纹图谱相似度评价系统(2004 A版)》(以下简称“相似度软件”)进行相似度评价,结果6次进样测得的指纹图谱相似度均大于0.9,表明仪器精密度良好,符合指纹图谱测定的要求。
2.4.2 重复性试验 取S5白茅根样品,按照“2.1”项下方法平行制备6份供试品溶液,按照“2.3”项下色谱条件测定,记录色谱图。经过相似度软件评价,6份样品测定的指纹图谱相似度均大于0.9,表明该方法重复性良好,符合指纹图谱测定要求。
2.4.3 稳定性试验 按照“2.1”项下方法制备S5白茅根供试品溶液1份,分别于0、4、8、12、24 h,按照“2.3”项下色谱条件进样测定,记录色谱图。经过相似度软件评价,不同时间进样测得的指纹图谱相似度均大于0.9,表明该供试品溶液在24 h内稳定。
2.4.4 完整性试验 按照“2.1”项下方法制备S5白茅根供试品溶液1份,按照“2.3”项下色谱条件进样测定,以流动相梯度最终浓度继续洗脱至170 min,采集色谱图,见图1。结果在采集110 min以后基本无色谱峰出现,表明采集110 min即可完整记录白茅根的化学成分信息。
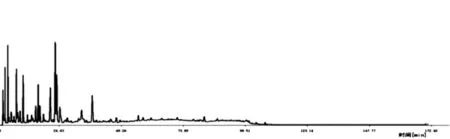
图1 完整性试验HPLC色谱
2.5 白茅根及炮制品指纹图谱建立
精密称取10个批次的鲜白茅根、白茅根与茅根炭药材,按照“2.1”项下方法,制备供试品溶液,按照“2.3”项下色谱条件依法测定,将记录的色谱图分别导入相似度软件,以每种白茅根样品的S5为参照谱,采用中位数法,时间窗宽度为0.5,多点校正,自动匹配,得到3种白茅根样品的共有模式及对照指纹图谱,见图2、图3、图4、图5、图6、图7。综合10个批次的3种白茅根样品,共标定23个共有峰,其中3种白茅根样品均含有共有峰13,且响应值高(茅根炭样品较低),基本上达到基线分离,故选择13号峰作为参照峰。从图谱可得,不同产地鲜白茅根和白茅根在化学成分组成上无明显变化,但其化学成分含量有所差异;茅根炭的化学成分类型及含量与鲜白茅根、白茅根都有明显差异,其中共有峰6的含量有了极大增加。

图2 10批鲜白茅根的HPLC叠加指纹图谱

图3 10批不同产地鲜白茅根对照指纹图谱

图4 10批白茅根的HPLC叠加指纹图谱

图5 10批不同产地白茅根对照指纹图谱

图6 10批茅根炭的HPLC叠加指纹图谱
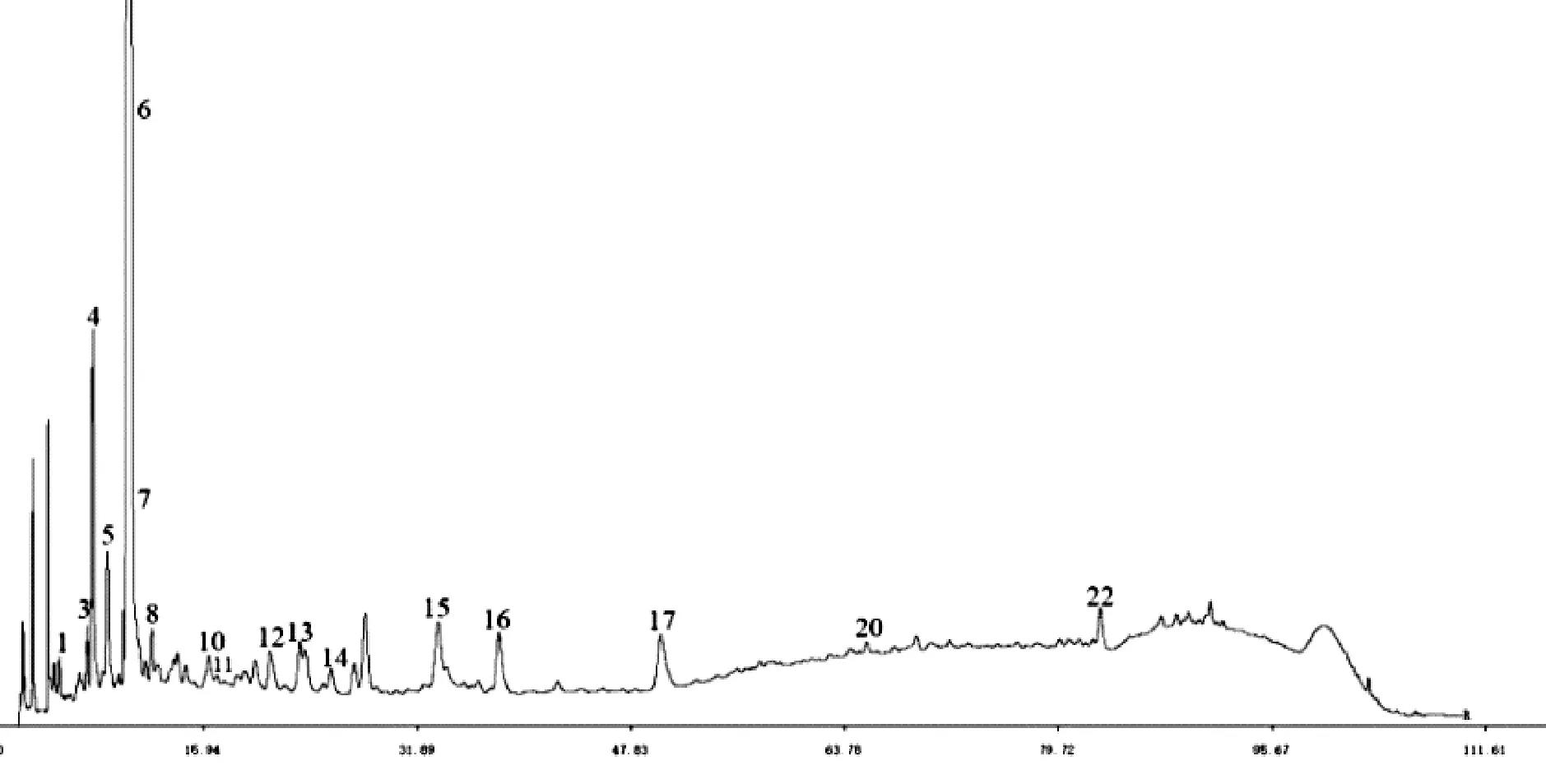
图7 10批不同产地茅根炭对照指纹图谱
2.6 共有峰的指认
精密吸取供试品溶液、对照品溶液各10 μL,按照“2.3”项下方法测定。结果表明供试品溶液色谱图中共有峰6、共有峰13分别与5-羟甲基糠醛对照品、绿原酸对照品色谱峰的保留时间和UV光谱图一致,可指认共有峰6为5-羟甲基糠醛,共有峰13为绿原酸。
2.7 白茅根指纹图谱相似度评价及化学计量学分析
2.7.1 白茅根指纹图谱相似度评价 将10个批次的白茅根样品色谱图(cdf.格式)全部导入相似度软件,以白茅根S5样品色谱图为参照谱,采用中位数法,时间窗宽度为0.5,多点校正,自动匹配后得到相似度分析结果。10个批次的白茅根与对照指纹图谱的相似度在0.432~0.985之间,其中样品S1的相似度颇低,可能是其采收季节不同于其他批次药材所致。除S1外,另外9个批次的白茅根与对照指纹图谱的相似度均大于0.82,表明同一采收期不同产地的白茅根药材在化学成分上保持一定的稳定性,见表3。

a:鲜白茅根;b:白茅根;c:绿原酸对照品;d:茅根炭;e:5-羟甲基糠醛对照品
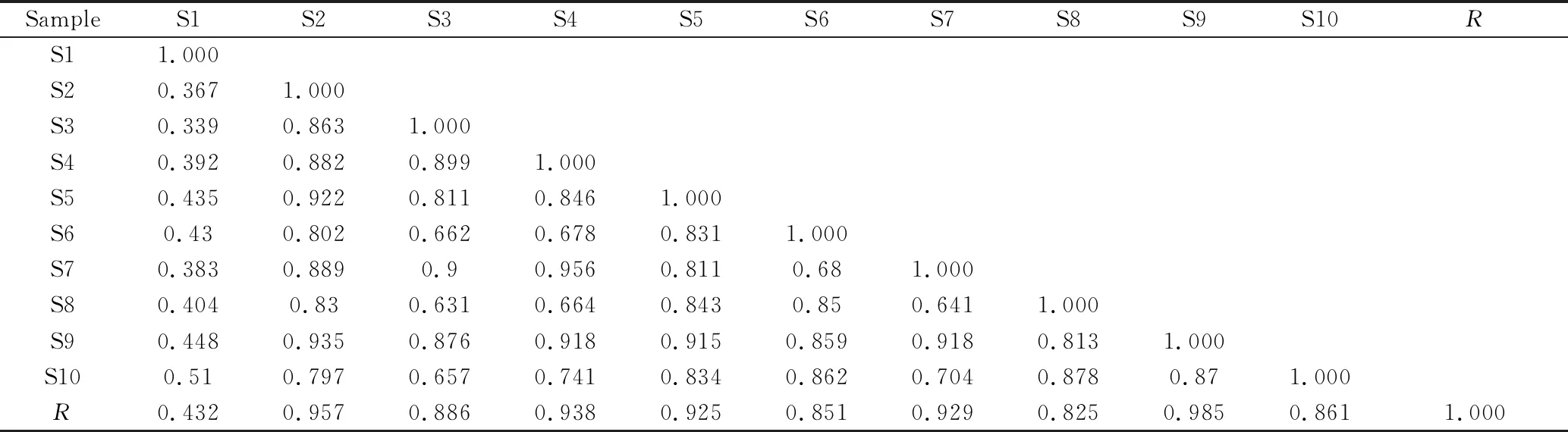
表3 白茅根样品的相似度结果
2.7.2 聚类分析(HCA) 将各个批次的白茅根共有峰峰面积进行Z-score标准化处理,导入Origin Pro 2018软件,采用组平均法进行聚类,利用欧氏距离( Euclidean distance)进行样品测定,结果当欧氏距离为7时,样品被分为两大类,S6被单独聚为一类,其他批次聚为一类;当欧氏距离为5时,样品被分为三大类,样品S5也被单独聚为一类;当欧氏距离为4时,样品被分为四大类,S7、S8、S10被分为一类,其余的被分为一类。

图9 10批白茅根样品聚类分析树状图
2.7.3 主成分分析(PCA) 将白茅根不同炮制品的30批共有峰峰面积数据导入Origin Pro 2018软件,以特征值及累计方差贡献率为依据进行主成分分析,见表4、图10。以特征值>1为提取标准,得到前4个主成分的累计方差贡献率为87.73%(>85%),说明提取的4个主成分可以代表原始色谱峰峰面积87.73%的信息,具有很好的代表性,提示主成分因子1、2、3、4可作为白茅根饮片的评价指标。

表4 特征值及方差贡献率

图10 主成分分析碎石图
主成分载荷矩阵可反映各变量对主成分的贡献大小及方向,根据主成分载荷矩阵结果可知,影响白茅根质量差异的是多成分联合作用的结果,见表5。第1主成分信息主要来源于色谱峰4、5、8、14、19、22,第2主成分信息主要来源于色谱峰7、10、12、13、11、14,第3主成分、第4主成分主要分别反映色谱峰10、18、20、23和1、11、17、18的化学信息。主成分1和2的累积贡献率达到67.5%,基本包含了白茅根指纹图谱的大部分信息,故以第1和2主成分得分建立分值图,可直观显示样品间的差异,见图11。由得分图可得,样品S5、S6分布均较为离散,S8、S10分布集中在一起,其他批次样品分布较为集中,此结果与聚类分析结果基本一致,两者相互得以印证。
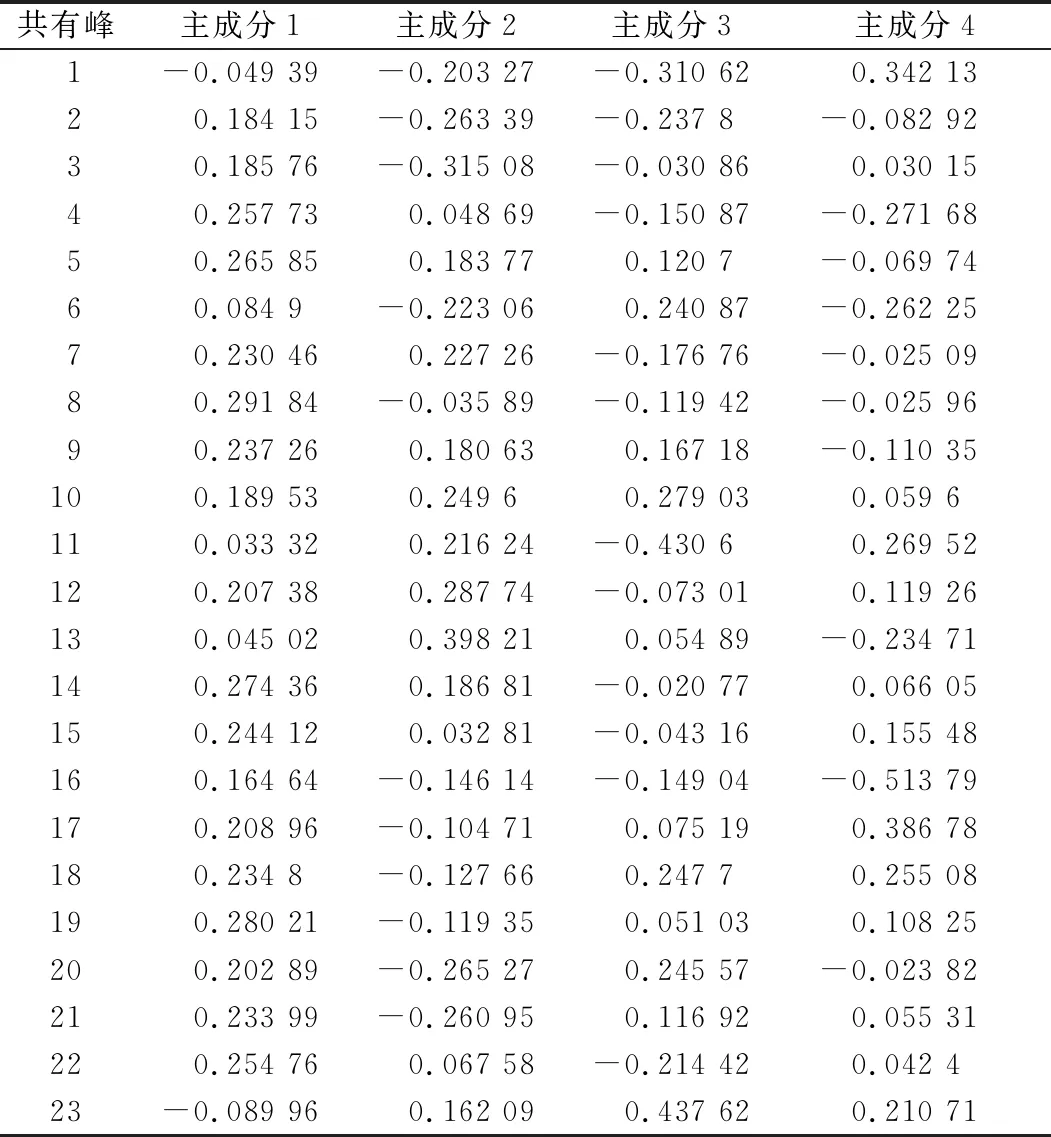
表5 主成分因子与变量间的相关系数

图11 主成分双标图(载荷图+分值图)
相似度分析、聚类分析和主成分分析的结果基本一致,10个批次的白茅根样品间在化学成分上保持一定的稳定性,但由于光照、水分、土壤等生境因素,彼此间也存在着差异,除S1(冬季购买)外,其他9批均为7月份采集,从分析结果推测样品在不同季节采收可能化学成分差异不大;样品S3是从山东购买所得,其余均为黔产,分析结果显示该批鲁产白茅根药材与黔产药材差异不大;样品S5、S9均采自同一个自治县,但分析结果显示两者差异较大,可能与其生长的小环境有关。可见,影响白茅根药材化学成分的因素很多,这对制定合理科学的质量控制标准尤为重要。
2.8 指纹图谱成分差异分析
2.8.1 指纹图谱色谱峰对比分析 通过比较鲜白茅根、白茅根、茅根炭的HPLC指纹图谱,鲜白茅根与白茅根之间的成分类型及含量差异不显著,而与白茅根与茅根炭相比其化学成分含量及类型都有一定变化。如图13示,在茅根炭指纹图谱中,Ⅰ、Ⅴ区域所代表的化学成分的含量均上升,其中Ⅰ区域中5-羟甲基糠醛含量显著增加,可能与其是由糖类成分在一定的温度、湿度、pH值等条件下发生脱水反应转化而来有关;而Ⅱ、Ⅲ、Ⅳ区域所代表的化合物含量在炒炭后均下降,其中Ⅲ区域所对应的化学成分为绿原酸类,含量下降最为明显,这可能与绿原酸热不稳定的性质有关,研究结果与文献基本一致。

g:白茅根HPLC指纹图谱;f:茅根炭HPLC指纹图谱
2.8.2 正交偏最小二乘判别分析(OPLS-DA) 为更好地观察组间差异,采用监督模式识别法对化学成分差异较大的白茅根和茅根炭进行两组间 OPLS-DA分析,绘制 OPLS-DA模型得分图,白茅根和茅根炭样品被分在不同的区域,详见图13。该OPLS-DA模型中,拟合参数模型和预测参数均大于0.5,表明所建模型较稳定且预测能力较强。S-plot图S曲线的每一个点代表一个化合物,差异性化合物分布在S型曲线的上下两端。普遍认为变量重要性投影值 (VIP值)较大的变量(一般>1),对分类的贡献越大[25]。选取位于S-plot图两端并且VIP>1的点作为区分白茅根和茅根炭的差异性化合物,在S-plot图中,色谱峰6、15、17处于坐标图内的最右上方,色谱峰13、9、12、2集中分布坐标图内的最左下方,且VIP值均>1,提示这些色谱峰所对应的化学成分对分类贡献较大,见图14、图15。该结果在统计分析角度进一步对白茅根和茅根炭指纹图谱对比分析进行验证,并筛选出炒炭前后差异性化合物,其中峰6和13被指认为5-羟甲基糠醛和绿原酸,其他5个色谱峰有待于进一步确认其结构。

图13 白茅根和茅根炭样品的OPLS-DA模型得分图

图14 白茅根和茅根炭样品的OPLS-DA模型S-plots图
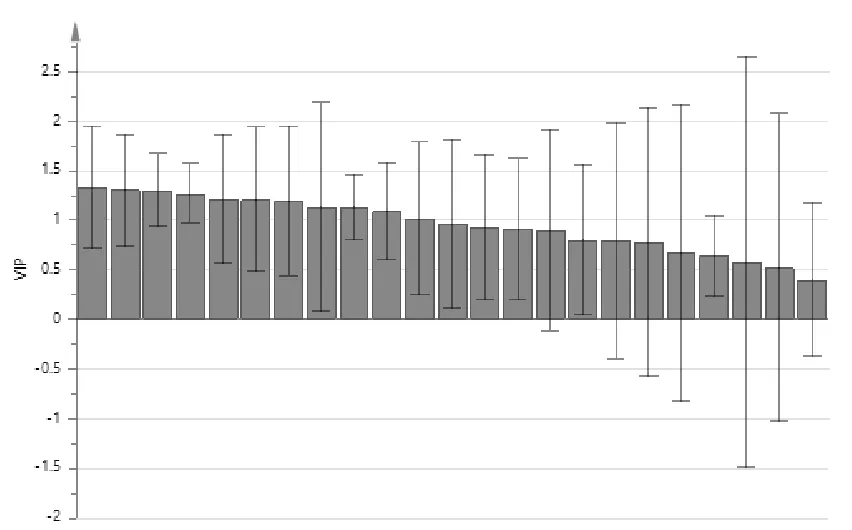
图15 OPLS-DA模型中共有峰的VIP值
3 讨论
本文前期对供试品溶液的提取条件进行了筛选,分别考察了提取方法(超声、回流)、提取溶剂(30%甲醇、50%甲醇、70%甲醇、90%甲醇、30%乙醇、50%乙醇、70%乙醇、90%乙醇)、料液比(1∶10、1∶20、1∶30、1∶40、1∶50)、提取时间(回流1 h、2 h、3 h、4 h)的影响,结果表明以70%乙醇为提取溶剂,料液比为1∶20,回流提取3 h时,提取较为完全,指纹图谱的峰容量较大,且各色谱峰的响应值均较高,基线平稳,故选择该条件。
本实验还考察了不同色谱柱Diamonsil C18(250 mm×4.6 mm,5 μm)、Dikma Platisil Dos C18(250 mm×4.6 mm,5 μm),流动相(乙腈-0.1%磷酸溶液、甲醇-乙腈-0.1%磷酸溶液、乙腈-0.05%磷酸水),检测波长(278 nm、280 nm、284 nm、323 nm),柱温(25 ℃、30 ℃),检测器(蒸发光散色检测器、紫外检测器)的影响。比较不同色谱条件下指纹图谱中各色谱峰的分离效果及其响应值大小,最终确定文中最佳色谱条件。
根据文献[26-27]报道,临床试验证明茅根炭的收敛止血之功远优于鲜茅根和白茅根,可能与5-羟甲基糠醛的积累相关,该成分究竟与收敛止血的功效有何相关作用,其收敛止血之功是否与茅根炭中新生成的化学成分有关需进一步通过药理试验进行验证。指纹图谱中化学成分类型及含量变化与白茅根与茅根炭的药理作用有何关系?对于不同炮制饮片的质量控制方法如何建立?以上问题值得我们深入研究。
综上所述,本文建立的指纹图谱测定方法准确可靠,简便易行,可较全面地反映白茅根炮制前后化学成分的变化情况,为白茅根药材和饮片的药效物质基础研究及其质量控制及评价提供了参考。
猜你喜欢
吉林中医药(2021年9期)2021-03-27 11:17:22
小哥白尼(趣味科学)(2021年11期)2021-02-28 08:35:00
小天使·一年级语数英综合(2020年10期)2020-12-16 02:57:11
妇女生活(2019年6期)2019-06-26 02:56:50
天然产物研究与开发(2019年1期)2019-03-01 05:41:08
中成药(2018年11期)2018-11-24 02:56:46
华声文萃(2018年3期)2018-08-04 19:03:18
中成药(2017年10期)2017-11-16 00:50:42
自动化学报(2016年8期)2016-04-16 03:39:00
青少年科技博览(中学版)(2015年7期)2015-08-12 18:50:24
