
新烟碱类杀虫剂环氧虫啶的合成工艺优化
2021-04-03 01:53王涛
世界农药 2021年3期
王 涛
(上海生农生化制品股份有限公司,上海 201108)
环氧虫啶[1]是由华东理工大学李忠团队创制,与上海生农生化制品股份有限公司共同开发并成功商品化的一种新烟碱类杀虫剂。环氧虫啶(图1)具有独特的顺式硝基构型和七元氧桥杂环结构,而吡虫啉等其他烟碱类杀虫剂中的硝基均为反式构型。环氧虫啶的作用机制与其他新烟碱类不同,与其他烟碱类杀虫剂不产生交互抗性问题,对吡虫啉等杀虫剂产生抗性的害虫有效[2]。因此,环氧虫啶既可有效地防治害虫,还可以解决昆虫抗性问题,同时具有良好的生物安全性[3-4]。环氧虫啶具有广阔的市场前景和很高的开发价值。
文献报道的环氧虫啶合成方法主要有以下几种:⑴ 以2-氯-5-氯甲基吡啶为原料,与过量的乙二胺缩合得到氯吡啶苄乙二胺;然后和1,1-二甲硫基-2-硝基乙烯发生环合反应脱除两分子甲硫醇气体,得到氯吡啶硝基亚甲基咪唑烷中间体;最后该中间体与2,5-二甲氧基四氢呋喃在酸性条件下关环得到环氧虫啶[1]。该合成方法收率低,并且会产生有毒和恶臭的甲硫醇,安全环保上存在较大隐患;⑵ 以偏二氯乙烯为原料,经硝化后再与乙二胺缩合得到硝基亚甲基咪唑烷;然后与2-氯-5-氯甲基吡啶在二甲基亚砜溶剂下发生缩合反应得到氯吡啶硝基亚甲基咪唑烷中间体;最后与丁二醛发生关环反应得到环氧虫啶[5]。该合成方法收率偏低,且使用了难回收的二甲基亚砜溶剂,工业化价值不高;⑶ 以偏二氯乙烯为原料,先经硝化后再在碱性条件下醇解得到二甲氧基硝基乙烯;然后与乙二胺缩合得到硝基亚甲基咪唑烷;再与2-氯-5-氯甲基吡啶在碱性条件下缩合得到氯吡啶硝基亚甲基咪唑烷中间体[6-7]。该合成方法虽然比方法2 收率有所提高,但是二甲氧基硝基乙烯稳定性较差,与2-氯-5-氯甲基吡啶缩合后溶剂仍然难回收,能耗高,不适合工业化。⑷ 仍以偏二氯乙烯为原料,经硝化、醇解得到二甲氧基硝基乙烯,然后与方法一中得到的氯吡啶苄乙二胺在水做溶剂中进行缩合反应,得到氯吡啶硝基亚甲基咪唑烷中间体[8]。该合成方法虽然避免了高沸点溶剂的使用,但仍需使用二甲氧基硝基乙烯,此物质稳定性较差,收率偏低,工业化难度高。
笔者在参考文献的基础上,以2-氯-5-氯甲基吡啶为原料,先与乙二胺缩合得到氯吡啶苄乙二胺,然后与偏二氯乙烯经硝化得到的1,1-二氯硝基乙烯进行缩合得到氯吡啶硝基亚甲基咪唑烷中间体,然后与丁二醛水溶液在酸性条件下发生关环反应得到环氧虫啶。
1 试验部分
1.1 仪器与试剂
仪器:布鲁克 400MHz核磁共振仪(瑞士Bruker公司);安捷伦 1260 液相色谱仪(美国Agilent 公司);LC-MS 液质联用仪(美国Agilent 公司);爱朗N-1100旋转蒸发仪(日本东京理化公司)。
试剂:2-氯-5-氯甲基吡啶(98%,上海阿拉丁生化科技有限公司);乙二胺、氢氧化钠、氢氧化钾、三氟乙酸等(AR,国药集团化学试剂有限公司);1,1-二氯硝基乙烯(97%,自制)[5];丁二醛水溶液(50%,自制)。
1.2 合成方法
环氧虫啶的合成路线如图2 所示。
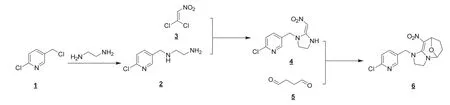
图2 环氧虫啶的合成路线
1.2.1 氯吡啶苄乙二胺(2)的合成
在装有回流冷凝管、温度计、恒压滴液漏斗和搅拌器的500 mL 四口烧瓶中加入54.5 g (0.90 mol)乙二胺、13.3 g (0.33 mol)氢氧化钠、150 mL 二氯甲烷,冰浴降温至5 ℃,滴加50.0 g (0.30 mol) 2-氯-5-氯甲基吡啶,加毕,5 ℃保温搅拌1 h,加入100 mL水水洗,静置分层,有机相减压浓缩回收溶剂,得到54.3 g 浅黄色油状液体,即为氯吡啶苄乙二胺,含量95.6% (HPLC),收率93.4%。LC-MS(m/z): 186 [M+1]+。
1.2.2 氯吡啶硝基亚甲基咪唑烷(4)的合成
在装有回流冷凝管、温度计、恒压滴液漏斗和搅拌器的500 mL 四口烧瓶中加入40.0 g (0.21 mol)氯吡啶苄乙二胺、200.0 g (0.50 mol)10%氢氧化钠水溶液、40 mL 二氯甲烷,冰浴降温至10 ℃,滴加36.6 g (0.25 mol) 1,1-二氯硝基乙烯,加毕,10 ℃保温搅拌30 min,反应物料过滤,滤饼水洗,烘干得到47.5 g浅黄色粉末状固体,即为氯吡啶硝基亚甲基咪唑烷,含量97.2% (HPLC),收率86.6%。MS-EI (m/z): 254 M+;1H NMR (400 MHz, DMSO-d6),δ:8.89(s, 1H), 8.37(s, 1H), 7.78(d, J = 8 Hz, 1H), 7.52(d,J = 8 Hz,1H), 6.76(s, 1H), 4.49(s, 2H), 3.64-3.51(m, 4H)。
1.2.3 环氧虫啶(6)的合成
在装有回流冷凝管、温度计和搅拌器的500 mL四口烧瓶中加入36.1 g (0.21 mol)50%丁二醛水溶液、26.5 g (0.27 mol)三氟乙酸、250 mL 二氯甲烷,控制温度20 ℃,分批加入50.0 g(0.19 mol)氯吡啶硝基亚甲基咪唑烷固体,加毕,20 ℃保温搅拌2 h,向反应液中滴加10%氢氧化钠水溶液使pH 至10 左右,静置分层,有机相减压浓缩回收溶剂,剩余物加入100 mL 甲醇打浆,过滤,烘干得到50.1 g 米白色粉末状固体,即为环氧虫啶,含量98.1%(HPLC),收率80.2%。LC-MS (m/z): 323 [M+1]+;1H NMR (400 MHz, CDCl3),δ: 8.26(s, 1H), 7.77(d, J = 12 Hz, 1H), 7.27(d, J = 8 Hz, 1H), 5.56(d, J = 4 Hz, 1H), 5.16(d, J = 16 Hz, 1H), 5.10(d, J = 4 Hz, 1H), 4.55(d, J = 16 Hz, 1H), 3.69-3.60(m, 3H), 3.47-3.39(m, 1H), 2.21-1.94(m, 4H)。
2 结果与讨论
2.1 乙二胺的量对氯吡啶苄乙二胺(2)收率的影响
反应条件同1.3.1 节,考察乙二胺的量对氯吡啶苄乙二胺(2)收率的影响,试验结果见表1。
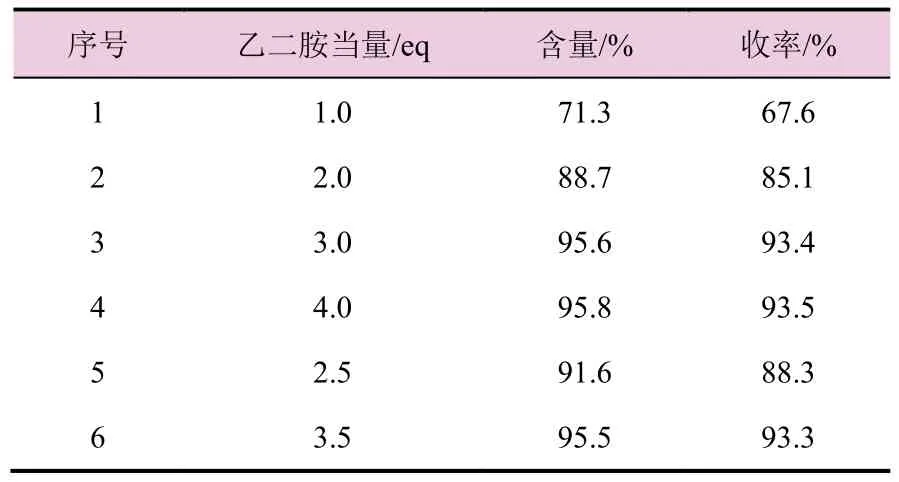
表1 乙二胺当量对氯吡啶苄乙二胺(2)收率的影响
由表1 数据可知,乙二胺当量过低,氯吡啶苄乙二胺含量和收率都明显下降,主要原因是乙二胺分子结构中有2 个胺基,都可以与2-氯-5-氯甲基吡啶反应形成双分子缩合的杂质。增加乙二胺当量,同时采用滴加2-氯-5-氯甲基吡啶,可有效降低双分子缩合杂质的产生,继续再增加乙二胺当量至4当量时,含量和收率没有明显增加。故选择乙二胺当量为3。
2.2 不同缚酸剂对氯吡啶苄乙二胺(2)收率的影响
反应条件同1.3.1 节,考察不同的缚酸剂对氯吡啶苄乙二胺(2)收率的影响,试验结果见表2。
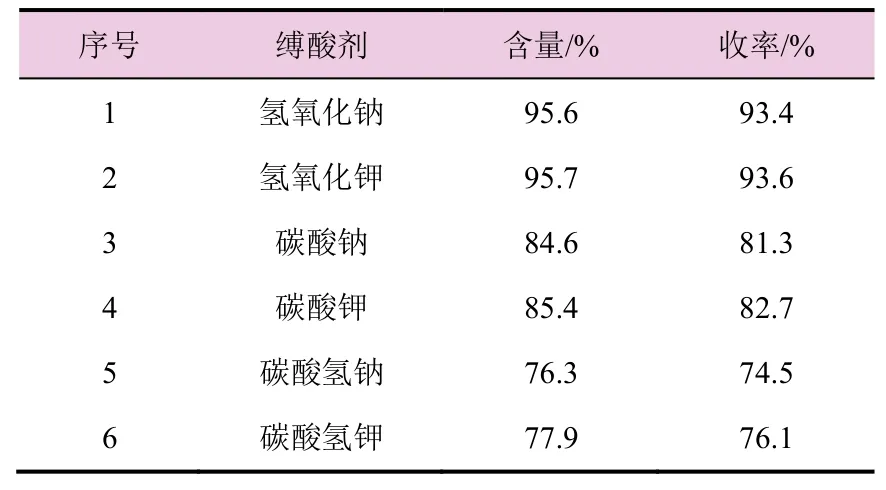
表2 不同缚酸剂对氯吡啶苄乙二胺(2)收率的影响
由表2 数据可知,随着缚酸剂碱性的减弱,氯吡啶苄乙二胺含量和收率也随着下降,而采用氢氧化钠和氢氧化钾做缚酸剂时,含量和收率都较高。考虑到原材料成本因素,故选择氢氧化钠作为缚酸剂。
2.3 反应温度对氯吡啶苄乙二胺(2)收率的影响
反应条件同1.3.1 节,考察不同的反应温度对氯吡啶苄乙二胺(2)收率的影响,试验结果见表3。
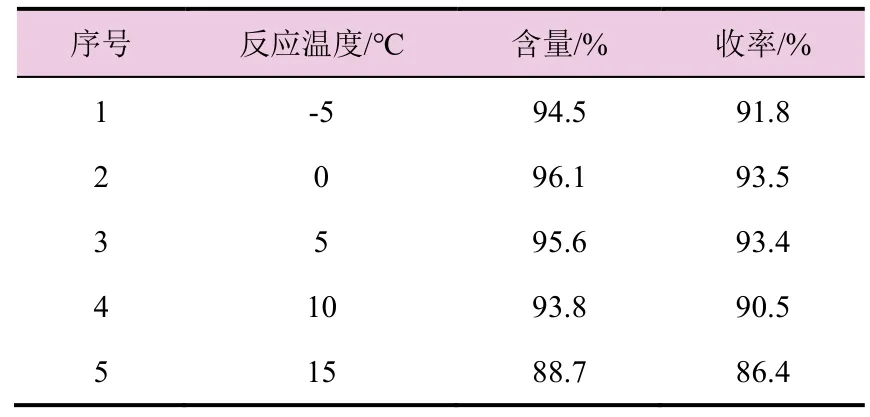
表3 反应温度对氯吡啶苄乙二胺(2)收率的影响
由表3 数据可知,反应温度过低,原料转化不完全,氯吡啶苄乙二胺收率有所下降,而提高反应温度,副反应增加,杂质变多,收率也明显下降。故选择适宜的反应温度为5 ℃。
2.4 物料配比对氯吡啶硝基亚甲基咪唑烷(4)收率的影响
反应条件同1.3.2 节,考察物料配比对氯吡啶硝基亚甲基咪唑烷(4)收率的影响,试验结果见表4。
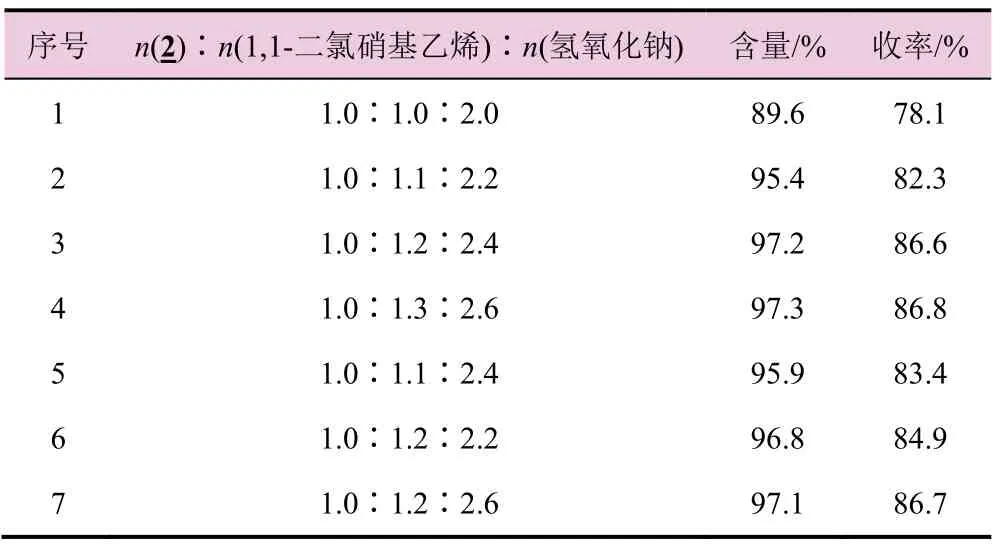
表4 物料配比对氯吡啶硝基亚甲基咪唑烷(4)收率的影响
由表4 可知,随着1,1-二氯硝基乙烯和碱的当量下降,氯吡啶苄乙二胺物料转化不完全,导致氯吡啶硝基亚甲基咪唑烷收率和含量明显下降;增加1,1-二氯硝基乙烯和碱的当量,收率和含量也没有增加。保持碱当量不变,减少1,1-二氯硝基乙烯的当量,收率也降低了;保持1,1-二氯硝基乙烯的当量不变,减少碱的当量,收率有所下降,增加碱的当量,收率也没有明显增加。故适宜的物料配比为n(2)∶n(1,1-二氯硝基乙烯)∶n(氢氧化钠)= 1.0∶1.2∶2.4。
2.5 反应温度对氯吡啶硝基亚甲基咪唑烷(4)收率的影响
反应条件同1.3.2 节,考察不同反应温度对氯吡啶硝基亚甲基咪唑烷(4)收率的影响,试验结果见表5。

表5 反应温度对氯吡啶硝基亚甲基咪唑烷(4)收率的影响
由表5 数据可知,降低反应温度,由于原料转化不完全,收率有所下降,而提高反应温度,副反应增加,收率也明显下降。故选择适宜的反应温度为10 ℃。
2.6 投料方式对关环反应收率的影响
反应条件同1.3.3 节,考察不同的投料方式对关环反应中环氧虫啶收率的影响,试验结果见表6。

表6 投料方式对关环反应收率的影响
由表6 数据可知,投料方式对环氧虫啶收率的影响较大,采用一锅投、滴加丁二醛或滴加三氟乙酸的投料方式,环氧虫啶的收率均偏低,可能的主要原因是中间体(4)局部浓度过高后,多分子中间体易与丁二醛两边的醛基加成生成杂质;而采用分批加中间体(4)或滴加(4)的溶液时,环氧虫啶的收率明显提高。考虑到投料方便操作和简化,故选择合适的投料方式为分批加氯吡啶硝基亚甲基咪唑烷固体(4)。
2.7 不同酸对关环反应收率的影响
反应条件同1.3.3 节,考察不同种类的酸对关环反应中环氧虫啶收率的影响,试验结果见表7。
由表7 数据可知,酸对关环反应收率的影响也较大,无机酸类的关环收率低于有机酸类的,在进一步考察的几个有机酸类中三氟乙酸的关环收率最高;而在关环反应的体系中,若不加任何酸,中间体(4)不反应,得不到环氧虫啶,故选择三氟乙酸作为关环反应的酸催化剂。
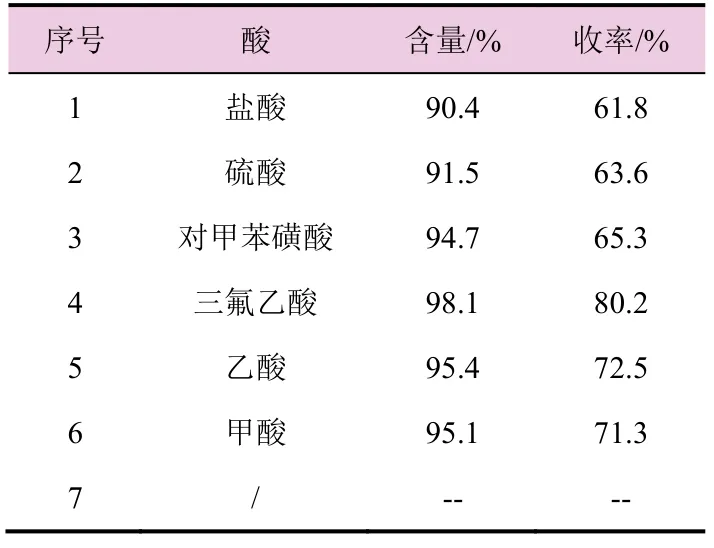
表7 不同的酸对关环反应收率的影响
2.8 关环反应合成环氧虫啶机理探讨
在关环反应合成环氧虫啶的反应体系中,氯吡啶硝基亚甲基咪唑烷和丁二醛在酸性条件下先加成得到含七元环的双羟基加成物,然后再在酸性条件下,双羟基醚化脱水得到含氧桥环的七元杂环结构。可能的反应机理如图3 所示。

图3 关环反应合成环氧虫啶可能的反应机理
3 结 论
以2-氯-5-氯甲基吡啶为原料,先与乙二胺缩合得到氯吡啶苄乙二胺,然后与1,1-二氯硝基乙烯进行缩合得到氯吡啶硝基亚甲基咪唑烷中间体(4),对两步缩合反应进行了工艺优化,以两步80.8% (以2-氯-5-氯甲基吡啶计)的收率得到中间体(4)。
环氧虫啶关环反应中,通过对投料方式和酸种类的优化以及反应机理的探讨,以80.2%的关环收率得到环氧虫啶。该合成方法具有路线短,工艺过程简单,各步收率较高等优点,适合工业化放大。
猜你喜欢
云南化工(2022年4期)2022-05-17
天津建设科技(2022年2期)2022-05-05
科学导报(2022年21期)2022-04-10
动物营养学报(2022年1期)2022-02-20
北京大学学报(自然科学版)(2019年6期)2019-11-27
火工品(2019年3期)2019-09-02
成长·读写月刊(2017年3期)2017-04-08
科技视界(2016年26期)2016-12-17
今日农药(2014年7期)2014-09-15
食品工业科技(2014年6期)2014-05-10
