
Congenital hepatic fibrosis in a young boy with congenital hypothyroidism: A case report
2021-03-30 08:09FangFeiXiaoYiZhongWangFangDongXiaoLuLiTingZhang
World Journal of Clinical Cases 2021年6期
Fang-Fei Xiao, Yi-Zhong Wang, Fang Dong, Xiao-Lu Li, Ting Zhang
Fan g-Fei Xiao, Yi-Zhong Wang, Fang Dong, Xiao-Lu Li, Ting Zhang, Department of Gastroenterology, Hepatology and Nutrition, Shanghai Children's Hospital, Shanghai Jiao Tong University, Shanghai 200062, China
Abstract BACKGROUND Congenital hepatic fibrosis (CHF) is a rare autosomal recessive disorder characterized by variable degrees of periportal fibrosis and malformation of bile ducts. CHF is generally accompanied by a variety of conditions or syndromes with other organ involvement.CASE SUMMARY We report a 5-year-4-month-old Chinese boy with congenital hypothyroidism(CH) diagnosed with CHF. The patient was diagnosed with CH by a newborn screening test and has since been taking levothyroxine. He has developed normally without neurocognitive deficits. Abnormal liver function was observed in the patient at the age of 4 years and 11 mo, and elevated levels of liver function indices were persistent for 5 mo. Radiological imaging indicated hepatosplenomegaly without narrowing of the portal vein but dilated splenic vein. A liver biopsy confirmed the pathological features of CHF. Genetic testing revealed two novel homozygous mutations, namely, c.2141-3T>C variant in PKHD1 related to CHF and c.2921G>A (p.R974H) in DUOX2 related to CH. The patient was treated with compound glycyrrhizin tablet, ursodeoxycholic acid, and levothyroxine after diagnosis. The patient achieved a favorable clinical outcome during a follow-up period of over 2 years.CONCLUSION Herein, we report the first case of a Chinese boy with comorbidity of CHF and CH, carrying both PKHD1 gene and DUOX2 gene novel mutations. Liver biopsy and genetic testing should be considered for the diagnosis of coexistent liver disease in CH patients with unexplained abnormal liver function.
Key Words: Congenital hepatic fibrosis; Congenital hypothyroidism; Liver biopsy;PKHD1; DUOX2; Case report; Genetic testing
INTRODUCTION
Congenital hepatic fibrosis (CHF) is a rare autosomal recessive hereditary disorder first described in 1961[1]. CHF is a fibropolycystic disease and is characterized by variable degrees of periportal fibrosis and malformation of bile ducts[2,3]. The prevalence of CHF is 1/10000-1/20000 in newborns[3,4]. CHF usually emerges in adolescence and early adulthood, and the clinical presentation of CHF varies greatly[4,5]. CHF has been reported to be accompanied by a broad range of disorders[5-10], but CHF in children with congenital hypothyroidism (CH) is extremely rare. CH is defined as a neonatal metabolic disorder, which may be caused by defects of thyroid hormone production in newborn infants[11]. Variants in different genes have been linked to CHF (such asPKHD1, TMEM67, andMKS1)[3]and CH (such asDUOX2,TSHR, andTPO)[12], but known variants do not account for all CHF or CH cases.
Herein, we report the case of a 5-year-4-month-old boy with CH and CHF, carrying bothPKHD1gene andDUOX2gene novel mutations. The clinical, laboratory,radiological, treatment and clinical outcomes as well as liver histologic characteristics and genetic variants of the patient are described in the present study.
CASE PRESENTATION
Chief complaints
A 5-year-4-month-old boy was admitted to our hospital due to persistent abnormal liver function for 5 mo.
History of present illness
Elevated levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST),alkaline phosphatase (ALP) and gamma-glutamyltransferase (GGT) were detected at the age of 4 years and 11 months, and these liver function indices were persistently abnormal for 5 mo (Table 1). During this period, the patient did not receive any relevant therapy.
History of past illness
After birth, he was diagnosed with CH by a newborn screening test, and he has been taking levothyroxine. No signs of persistent jaundice or hypoglycemia were found during the neonatal period.
Personal and family history
The patient was bornviacesarean section from healthy nonconsanguineous parents at full-term and weighed 2900 g.
Physical examination
He has developed normally (22 kg/117 cm) without neurocognitive deficits. Physicalexamination found that his spleen was palpable 2 cm below the left costal margin, and the liver edge was not palpable. Neuro-ophthalmic examination and eye movement were normal.
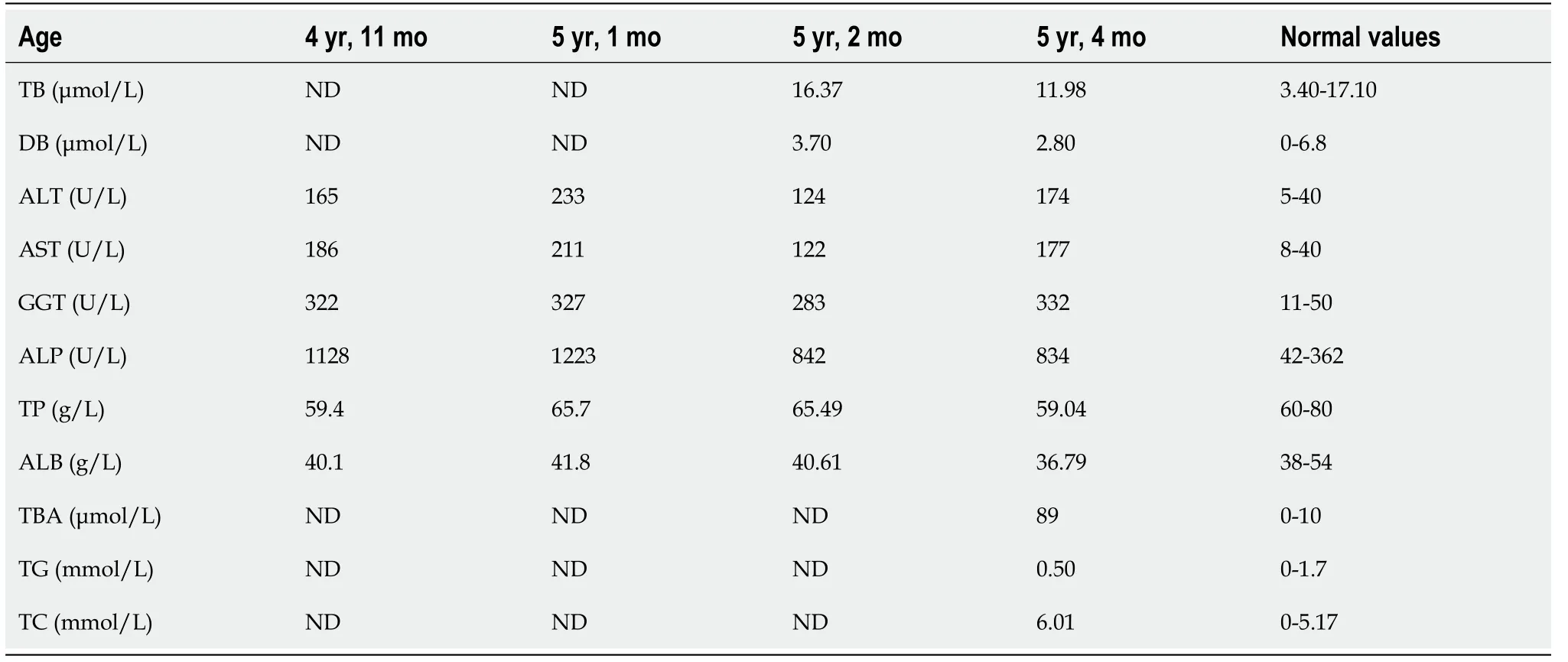
Table 1 Patient’s liver function laboratory data
Laboratory examinations
The liver biochemical profile on admission revealed elevated ALT 174 U/L (5-40), AST 177 U/L (8-40), GGT 332 U/L (11-50), ALP 834 U/L (42-362), total serum bile acid(TBA) 89 μmol/L (0-10), and total cholesterol 6.01 mmol/L (0-5.17), while total serum bilirubin, direct bilirubin and albumin were nearly normal (Table 1). Renal function and blood tests were normal. Blood coagulation function, alpha fetoprotein,ceruloplasmin, trace elements, lymphocyte subsets analysis, serum amino acid chromatography analysis, and urinary gas chromatography-mass spectrometry analysis were also normal. Tests for IgM antibodies to toxoplasma, rubella virus,cytomegalovirus, herpes simplex virus, Epstein-Barr virus, and human parvovirus B19 were negative. Hepatitis B surface antigen and anti-hepatitis C virus antibodies were negative.
Imaging examinations
Cardiac ultrasound and chest X-ray showed no abnormalities in the heart and lungs.Ultrasound of the abdomen revealed an enlarged liver with diffuse increased splenomegaly without cystic changes of the liver or kidneys. Magnetic resonance imaging (MRI) and computed tomography angiography (CTA) indicated hepatosplenomegaly and dilated splenic vein but without bile duct malformation and narrowing of portal vein.
Further diagnostic work-up
Liver biopsy demonstrated markedly enlarged portal tracts, widened fibrous septa,and proliferating bile ducts, which are typical of hepatic fibrosis (Figure 1A-D). Due to CH in this patient coexisting with unexplained liver fibrosis, clinical genome and exome sequencing was performed after obtaining informed consent from the parents.The following two novel homozygous mutations were identified: c.2141-3T>C in thePKHD1gene and c.2921G>A in theDUOX2gene (Figure 2A and B). In silico analysis revealed that the c.2141-3T>C mutation is predicted to destroy splice sites, influencing mRNA transcripts ofPKHD1and that the c.2921G>A homozygous variant in exon 22 ofDUOX2gene leads to a p.Arg974His substitution, which is predicted to affect protein splicing. Both mutations have been defined as uncertain significance according to the American College of Medical Genetics and Genomics guidelines (ACMGG), and they have not been described previously. The results also revealed that his parents were heterozygous carriers for both variants (Figure 2C-F).
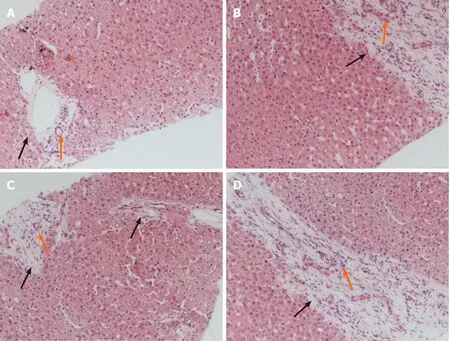
Figure 1 Histologic biopsy of the patient’s liver showing congenital hepatic fibrosis. A: The markedly enlarged portal tracts (black fork); B-D:Widened fibrous septa (black forks); A-D: Proliferating bile ducts (orange forks) (hematoxylin and eosin stain, × 100).
FINAL DIAGNOSIS
The diagnosis of CH with CHF was made.
TREATMENT
The patient was treated with compound glycyrrhizin tablet (25 mg twice daily),ursodeoxycholic acid (0.25 g/d), and levothyroxine (37.5 μg/d).
OUTCOME AND FOLLOW-UP
During a follow-up period of over 2 years, the transaminase values were reduced, and hepatic function gradually ameliorated (Figure 3). The compound glycyrrhizin tablet was discontinued at 15 mo after hospital discharge. Currently, the patient is generally in good health despite the level of TBA being slightly elevated, and he continues to take ursodeoxycholic acid and levothyroxine (Figure 3).
DISCUSSION
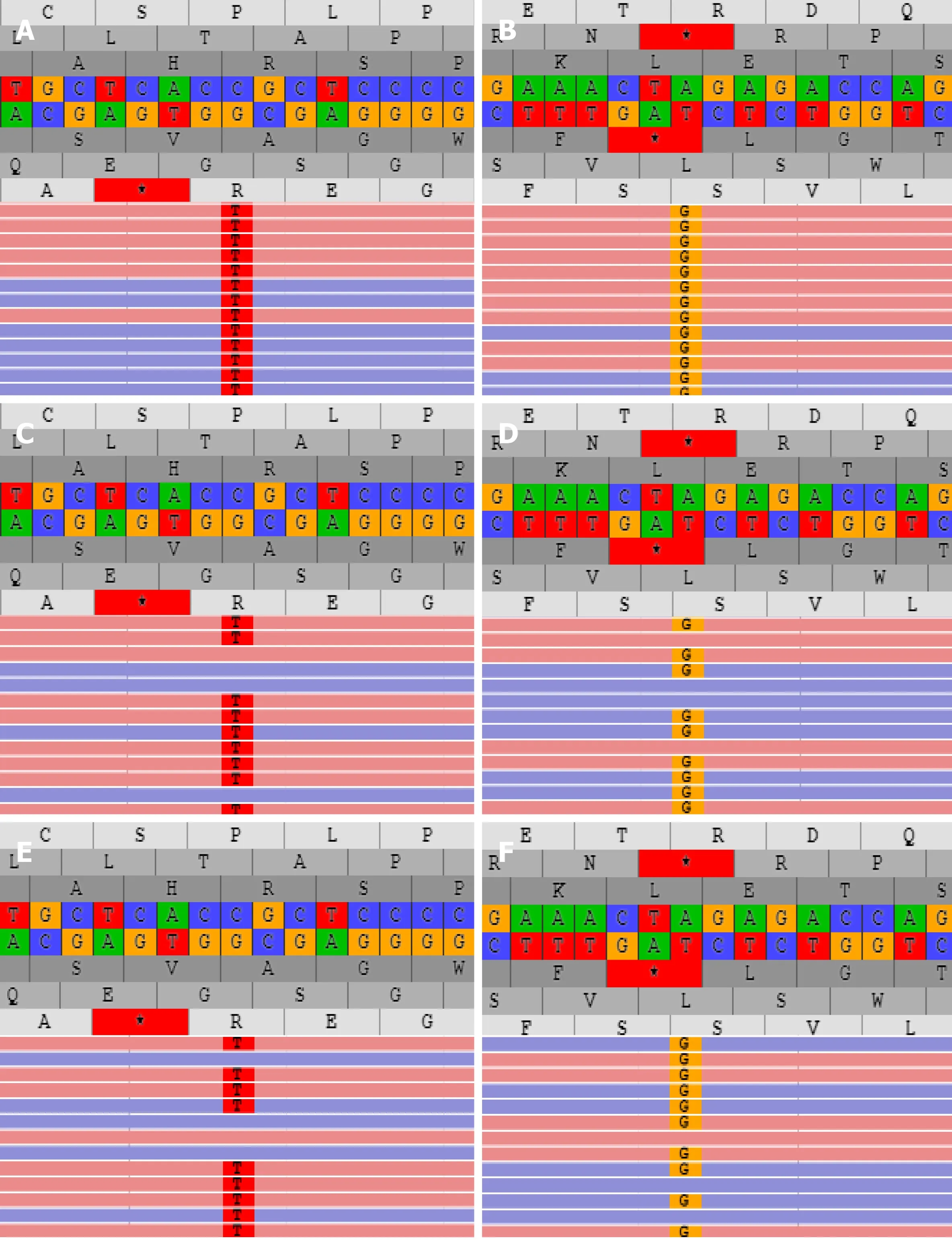
Figure 2 PKHD1 and DUOX2 mutations analysis of the patient and his family. A and B: The proband carried the homozygous mutation c.2141-3T>C of PKHD1 (A) and c.2921G>A of DUOX2 gene (B); C and D: The father had heterozygous c.2141-3T>C of PKHD1 (C) and c.2921G>A of DUOX2 gene (D); E and F:The mother carried heterozygous c.2141-3T>C of PKHD1 (E) and c.2921G>A of DUOX2 gene (F).
CHF is histologically defined as ductal plate malformation, abnormal proliferation of bile ducts, and progressive periportal fibrosis[2,13]. This disorder belongs to fibropolycystic diseases, and it involves damage to multiple organs[3]. Several syndromes associated with CHF have been reported, including Caroli disease[6,10],polycystic kidney disease (PKD)[7,14], and Prader-Willi syndrome[9]. CHF patients may present diverse clinical symptoms and even remain asymptomatic for a remarkably long period of time[10]. The common symptoms of CHF include hepatosplenomegaly,hypersplenism, gastroesophageal varices, and upper gastrointestinal bleeding caused by portal hypertension[3,4,10]. In this case, the patient experienced a period of mildly elevated liver enzymes with unknown etiology and splenomegaly. Based on an analysis of laboratory parameters and CTA, he was not diagnosed with portal hypertension. This required differential diagnosis as mild elevation of ALT and AST can occur in almost every liver disease[15]. Common liver diseases in children mainly include viral hepatitis, glycogen storage disease and Wilson disease[16]. In our case,hepatitis B surface antigen, anti-hepatitis C virus antibodies, ceruloplasmin and the fasting blood glucose test were normal. Abnormal GGT levels also required a separate differential diagnosis to exclude clinically relevant bile duct disease, such as primary sclerosing cholangitis and Caroli disease. The results of MRI did not show bile duct malformation; therefore, we ruled out these diseases. It is necessary to perform a comprehensive radiological examination in the diagnosis and differential diagnosis of CHF, and to assess the involvement of other organs. Moreover, the combination of CTA and MRI can provide more detailed information on the hepatosplenic artery and bile duct.
To date, the comorbidity of CHF in pediatric patients with CH is rarely reported in the literature. In 2003, Minamiet al[8]reported a 7-year-old girl with septo-optic dysplasia and CHF. She was diagnosed with growth hormone deficiency and hypothyroidism confirmed by low levels of peripheral thyroid hormones. In this report, we described a boy with CH coexisting with CHF that does not fit with the well-known syndromes associated with CHF. The patient had liver fibrosis and hepatosplenomegaly, and a liver biopsy confirmed the pathological features of CHF.To the best of our knowledge, this is the first reported case of CHF with CH from China. For children with CHF, no specific therapies are available to prevent or reverse the process of hepatic fibrosis. Therefore, periodic monitoring of liver fibrosis progression is important. FibroScan is a point-of-care and noninvasive clinical device to detect liver fibrosis, and is more sensitive than conventional ultrasound[17,18].Unfortunately, this test was not performed in our patient. CHF treatments depend on illness severity and complications. Liver transplantation is commonly one of the most effective treatments for children with CHF who develop severe complications of portal hypertension[4,5].

Figure 3 Patient’s liver function indices approximately 2 years after follow-up. The patient’s increased transaminases resolved a year after hospitalization, and the compound glycyrrhizin tablet was discontinued at 15 mo after hospital discharge. Albumin remained within normal levels, despite the level of total serum bile acid being slightly elevated. ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; ALB: Albumin; TBA: Total serum bile acid.
Several different genetic etiologies have been linked to CHF in the past decades,including mutations inPKHD1. ThePKHD1gene on chromosome 6p21.1-p12 encodes fibrocystin/polyductin, a protein highly expressed on kidney tubular epithelial cells and cholangiocytes of bile ducts[2,14]. This gene regulates cholangiocyte secretory activity and the expression of proteins involved in cell differentiation[2]. Researchers have reported over 300 mutations of thePKHD1gene[14]. Mutations of thePKHD1gene can cause defects in biliary cells and chronic low-grade inflammation derived from impaired bile ducts. The process triggers recruitment of macrophages at sites of injury,excessive deposition of collagen fibers, progressive fibrosis around the portal vein,compression of the portal vein radicles, and finally the development of several different syndromes, such as PKD[13]. Genetic testing in our patient revealed a novel homozygous mutation, c.2141-3T>C variant inPKHD1, which has been predicted to destroy splice sites, influencing mRNA transcripts ofPKHD1. However, the renal function test in this patient was normal, and there were no signs that the kidneys were affected.
CH is a neonatal metabolic disorder, which is defined by inadequate thyroid hormone production in newborn infants[11]. Patients with severe CH may suffer growth retardation and permanent intellectual disability without treatment. Molecular mechanism studies have demonstrated that mutations in genes involved in thyroid hormone synthesis are linked to CH, includingDUOX2[19].DUOX2is a member of the NAD phosphate oxidase (NOX/DUOX) family[20], and it generates H2O2and participates in theTPO-mediated iodide organification reaction, which is required for thyroid hormone biosynthesis. Variants in theDUOX2gene may result in transient or permanent forms of CH[20]. It has been reported that genetic mutations inDUOX2are the main genetic cause of CH in the Chinese population[21,22]. In our reported case, the novel homozygous mutation, c.2921G>A (p.R974H), inDUOX2was identified.
CONCLUSION
In summary, CHF is a rare hereditary disease, and the genotype-phenotype relationship has not been fully established. We report a rare comorbidity of CH and CHF with double novel homozygous mutations inPKHD1andDUOX2. Liver biopsy and genetic testing should be considered for the diagnosis of coexistent liver disease in CH patients with unexplained abnormal liver function.
ACKNOWLEDGEMENTS
The authors thank the family for participating and supporting this study.
 World Journal of Clinical Cases2021年6期
World Journal of Clinical Cases2021年6期
- World Journal of Clinical Cases的其它文章
- Interactive platform for peer review: A proposal to improve the current peer review system
- Animal models of cathartic colon
- New indicators in evaluation of hemolysis, elevated liver enzymes,and low platelet syndrome: A case-control study
- Analysis of hospitalization costs related to fall injuries in elderly patients
- Effect of alprostadil in the treatment of intensive care unit patients with acute renal injury
- Etomidate vs propofol in coronary heart disease patients undergoing major noncardiac surgery: A randomized clinical trial