
基于纳米孔全长转录组数据完善东方蜜蜂微孢子虫的基因组注释
2021-03-25 06:52陈华枝范元婵蒋海宾王杰范小雪祝智威隆琦蔡宗兵郑燕珍付中民徐国钧陈大福郭睿
中国农业科学 2021年6期
陈华枝,范元婵,蒋海宾,王杰,范小雪,祝智威,隆琦,蔡宗兵,郑燕珍,付中民,2,徐国钧,陈大福,2,郭睿,2
基于纳米孔全长转录组数据完善东方蜜蜂微孢子虫的基因组注释
陈华枝1,范元婵1,蒋海宾1,王杰1,范小雪1,祝智威1,隆琦1,蔡宗兵1,郑燕珍1,付中民1,2,徐国钧1,陈大福1,2,郭睿1,2
1福建农林大学动物科学学院(蜂学学院),福州 350002;2福建农林大学蜂疗研究所,福州 350002
【】利用已获得的纳米孔全长转录组数据对现有的东方蜜蜂微孢子虫()参考基因组的基因序列和功能注释进行完善。采用TransDecoder软件预测东方蜜蜂微孢子虫基因的开放阅读框(open reading frame,ORF)及相应的氨基酸。利用gffcompare软件将全长转录本与参考基因组注释的转录本进行比较,对基因组注释基因的非编码区向上游或下游延伸,修正基因的边界。利用MISA软件鉴定长度在500 bp以上的全长转录本的简单重复序列(simple sequence repeat,SSR)位点,包括单核苷酸重复、双核苷酸重复、三核苷酸重复、四核苷酸重复、五核苷酸重复、六核苷酸重复、混合SSR等类型。通过Blast工具将鉴定到的新基因和新转录本比对Nr、KOG、eggNOG、GO和KEGG数据库,从而获得功能注释。共预测出2 353个完整ORF,其中长度分布在0—100个氨基酸的ORF最多,占总ORF数的72.12%。共对东方蜜蜂微孢子虫的2 340个基因进行了结构优化,其中5′端延长的基因有1 182个,3′端延长的基因有1 158个。共鉴定到1 658个SSR,其中单核苷酸重复、双核苷酸重复、三核苷酸重复、四核苷酸重复的数量分别为1 622、23、7和6个;单核苷酸重复类型的SSR密度最大,达到182.32个/Mb,其次为混合SSR、双核苷酸重复和三核苷酸重复,分别达到6.90、2.78和0.73个/Mb。共鉴定出954个新基因,其中分别有951、333、371、422和321个新基因可注释到Nr、KOG、eggNOG、GO和KEGG数据库。此外,还鉴定出6 164条新转录本,其中分别有6 141、2 808、2 932、3 196和2 585条新转录本可注释到Nr、KOG、eggNOG、GO和KEGG数据库。新基因和新转录本注释数量最多的物种均为东方蜜蜂微孢子虫,其次是蜜蜂微孢子虫()。研究结果较好地完善了现有的东方蜜蜂微孢子虫参考基因组已注释基因的序列和功能注释,并补充和注释了大量参考基因组未注释的新基因和新转录本。
纳米孔测序;全长转录本;转录组;基因组;蜜蜂;东方蜜蜂微孢子虫
0 引言
【研究意义】东方蜜蜂微孢子虫()是细胞内寄生的单细胞真菌,特异性侵染成年蜜蜂的中肠上皮细胞,对蜜蜂幼虫也具有侵染性。目前,由于成熟的转基因操作技术平台缺失和公共数据库中微孢子虫基因注释信息的匮乏,东方蜜蜂微孢子虫的基因组注释很不完善。利用全长转录组数据对东方蜜蜂微孢子虫的完整开放阅读框(open reading frame,ORF)、简单重复序列(simple sequence repeat,SSR)及未注释基因和转录本进行鉴定,对已注释基因进行结构优化,可丰富和完善东方蜜蜂微孢子虫的参考基因组注释,为后续的生物信息学分析和分子生物学研究提供可靠的参考信息,也能为其他物种的基因组注释信息完善提供思路和方法借鉴。【前人研究进展】第一代测序技术即Sanger测序技术具有准确性高的优点,过去被成功用于人类[1]、家蚕()[2]和西方蜜蜂()[3]等物种的基因组测序和组装,但由于高成本和低通量的限制,逐渐被基于边合成边测序原理的第二代测序技术取代。近十几年来,以Illumina为代表的二代测序技术凭借高通量和成本持续下降的优势,在动物[4-5]、植物[6-7]和微生物[8-9]的基因组测序方面得到广泛应用,较大幅度地提升了物种的基因组组装质量。但二代测序具有GC偏好性且测序读段较短(不超过300 bp),需要通过生物信息学方法对短读段进行拼接,在测定重复序列方面劣势明显[10]。目前,除人类、小鼠和果蝇()等极少数模式物种的基因组组装到染色体水平外,绝大多数物种的基因组仅组装到contig或scafford水平,而且不同物种的基因组质量参差不齐[11-13]。近年来,随着以PacBio单分子实时(single- molecule real-time,SMRT)测序技术和Oxford纳米孔(nanopore)长读段测序技术为代表的三代测序技术的兴起与应用,人们通过纯三代测序或三代测序结合二代测序将越来越多物种基因组组装到染色体水平[14-16]。然而,目前三代测序的成本依然高昂,对于一些基因组较大的物种或经费有限的实验室,利用三代测序技术进行基因组测序还存在较大的困难。相对于基因组测序,利用三代测序技术进行转录组测序的成本较低且周期较短。利用PacBio SMRT测序数据完善小麦()和锡兰勾虫()基因组注释的研究已见报道[11,17]。Cornman等[18]通过454焦磷酸测序技术对东方蜜蜂微孢子虫的纯净孢子进行测序,利用CABOG软件拼接出5 465条contig,组装的基因组(assembly ASM18298v1)大小为7.86 Mb,GC含量为25.3%,包含2 060个蛋白编码基因。2015年,Pelin等[8]利用Illumina HiSeq技术重新测序并组装了东方蜜蜂微孢子虫的基因组(assembly ASM98816v1),其大小为8.82 Mb,包含110条contig,目前为NCBI Genome数据库推荐的参考基因组版本。但上述两个东方蜜蜂微孢子虫的基因组版本都只组装到contig水平,远未达到染色体水平,因而会对基于这两个基因组版本的生物信息学分析产生影响。因此,通过多组学数据对东方蜜蜂微孢子虫的基因序列和功能注释进行补充和完善尤为必要。【本研究切入点】前期研究中,笔者所在团队已利用Oxford nanopore测序技术对东方蜜蜂微孢子虫的纯净孢子进行测序,基于高质量的测序数据构建和注释了东方蜜蜂微孢子虫的首个全长转录组[19];并对东方蜜蜂微孢子虫基因的可变剪接和可变多聚腺苷酸化进行了系统鉴定和分析[20]。【拟解决的关键问题】利用已获得的全长转录组数据对东方蜜蜂微孢子虫参考基因组的完整ORF进行预测,对已注释基因进行结构优化,对未注释的SSR进行挖掘,并对未注释的新基因和新转录本进行鉴定和功能注释。
1 材料与方法
试验于2019年在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。
1.1 全长转录组数据来源
前期研究中,笔者所在团队利用Oxford Nanopore测序技术对东方蜜蜂微孢子虫的纯净孢子进行测序,获得了高质量的全长转录组数据,共测得6 988 795条原始读段(raw reads),居中长度(N50)、平均读长和最大读长分别为971、881和96 051 bp,共鉴定出10 243条非冗余全长转录本,N50、平均长度和最大长度分别为1 042、894和4 855 bp[19]。高质量的全长转录组数据可为本研究中的完整ORF预测、已注释基因的结构优化、SSR位点鉴定与分析、新基因鉴定与功能注释,以及新转录本的鉴定和功能注释提供可靠的数据支撑。
1.2 基因结构优化
由于软件和数据本身的局限性,导致多数基因组的基因结构信息不够精确,需要进一步优化。利用gffcompare软件将本研究鉴定到的全长转录本与东方蜜蜂微孢子虫参考基因组注释的转录本进行比较,对基因组注释的基因结构信息进行补充。如果在注释基因边界之外的区域有比对上的读段(mapped reads)支持,则将基因的UTR向上游或下游延伸,修正基因的边界。
1.3 SSR位点的鉴定及分析
MISA(MIcroSAtellite identification tool)软件[21]可以通过对转录本序列的分析,鉴定出7种类型的SSR,包括单核苷酸重复(p1)、双核苷酸重复(p2)、三核苷酸重复(p3)、四核苷酸重复(p4)、五核苷酸重复(p5)、六核苷酸重复(p6)、混合SSR(c,即两个SSR之间的距离<100 bp)。从去冗余的全长转录本中筛选长度在500 bp以上的全长转录本,利用MISA软件预测SSR位点,采用默认参数。
1.4 新基因和新转录本的鉴定及数据库注释
以东方蜜蜂微孢子虫参考基因组(assembly ASM98816v1)[8](gff文件)和本研究中去冗余后的全长转录本文件为基础,获得一个数据格式与注释文件相同的gff文件,利用gffcompare软件将2个gff文件进行比较,对于在参考基因组上没有注释信息的基因和转录本,将其定义为新基因和新转录本。利用Blast工具将上述新基因和新转录本分别比对Nr、KOG、eggNOG、GO和KEGG数据库,从而获得相应的功能注释。
1.5 ORF预测
TransDecoder(v3.0.0)软件可基于ORF长度、对数似然函数值、氨基酸序列与Pfam数据库蛋白质结构域序列的比对等信息,从转录本序列中识别可靠的潜在编码区序列(coding sequence,CDS)。采用TransDecoder(v3.0.0)软件对上述新转录本的CDS及其对应氨基酸序列进行识别,从而预测ORF。同时预测到起始密码子和终止密码子的ORF为完整ORF。
2 结果
2.1 东方蜜蜂微孢子虫的基因结构优化
共对东方蜜蜂微孢子虫的2 340个基因的结构进行优化,其中5′端延长的基因有1 182个,3′端延长的基因有1 158个。部分基因的结构优化信息详见表1。
2.2 东方蜜蜂微孢子虫的SSR鉴定及分析
如表2所示,在8 265 494 bp的序列中共鉴定到1 658个SSR,含有SSR超过1个的基因数为212个,以混合物形式存在的SSR有65个;此外,单核苷酸重复、双核苷酸重复、三核苷酸重复、四核苷酸重复的数量分别为1 622、23、7和6个。进一步分析SSR的类型分布,结果显示p1类型的SSR密度最大,达到182.32个/Mb,其次为c、p2和p3,分别达到6.90、2.78和0.73个/Mb。
2.3 东方蜜蜂微孢子虫的新基因鉴定及数据库注释
共鉴定出954个新基因,其中分别有951、333、371、422和321个新基因可注释到Nr、KOG、eggNOG、GO和KEGG数据库。新基因注释数量最多的物种是东方蜜蜂微孢子虫(798),其次为蜜蜂微孢子虫()(55)和按蚊微孢子虫()(43)(图1-A)。新基因可注释到KOG数据库的25个功能类别,包括翻译后修饰、蛋白折叠和分子伴侣(43),转录(41),一般功能预测(41),复制、重组和修复(39),以及翻译、核糖体结构和生物合成(36)等(图1-B)。此外,新基因可注释到eggNOG数据库的25个功能类别,包括未知功能(94),翻译、核糖体结构和生物合成(48),复制、重组和修复(47),翻译后修饰、蛋白折叠和分子伴侣(36),以及转录(35)等(图1-C)。括号内的数字代表注释上的新基因数量。
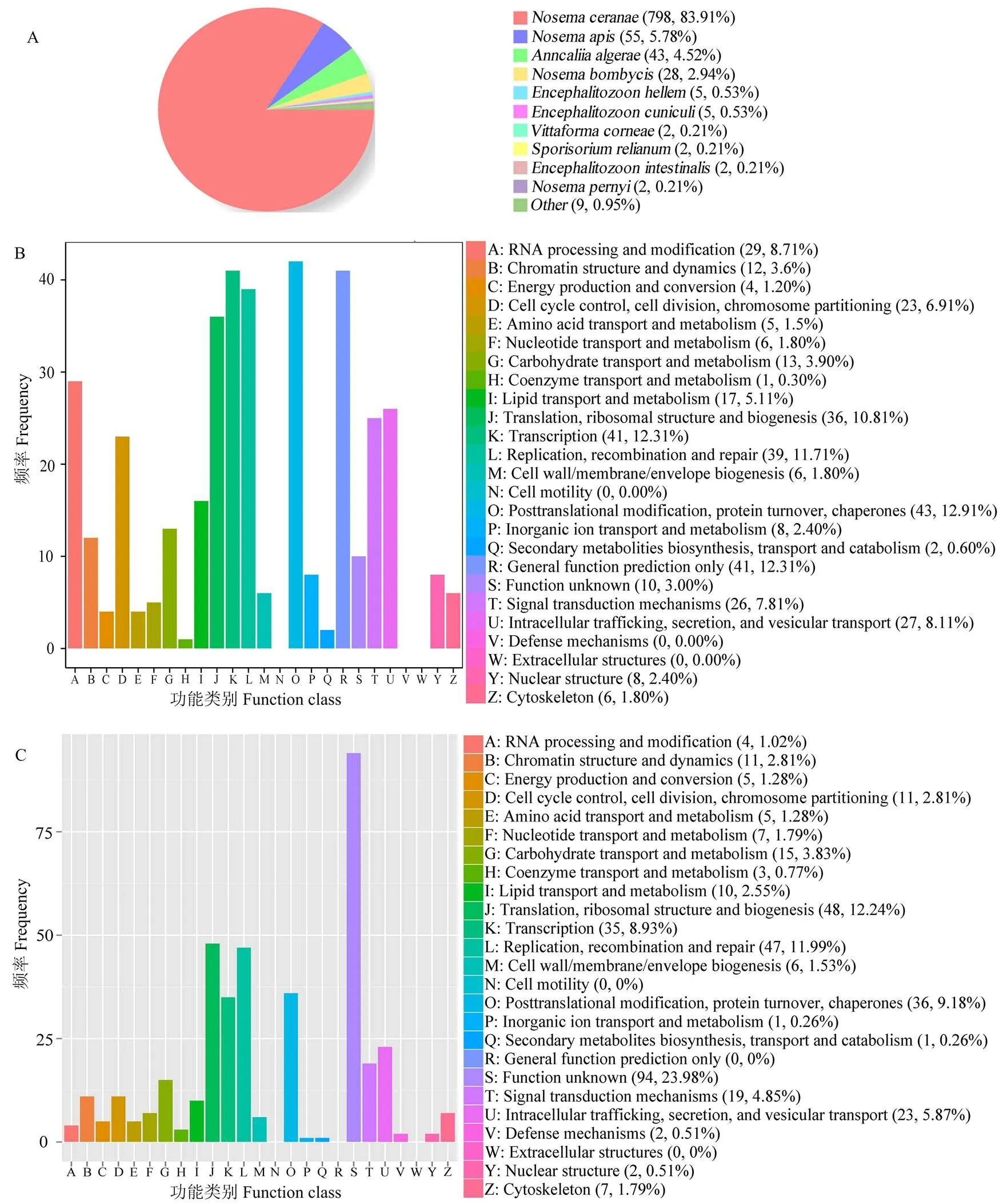
图1 东方蜜蜂微孢子虫新基因的Nr(A)、KOG(B)和eggNOG(C)数据库注释
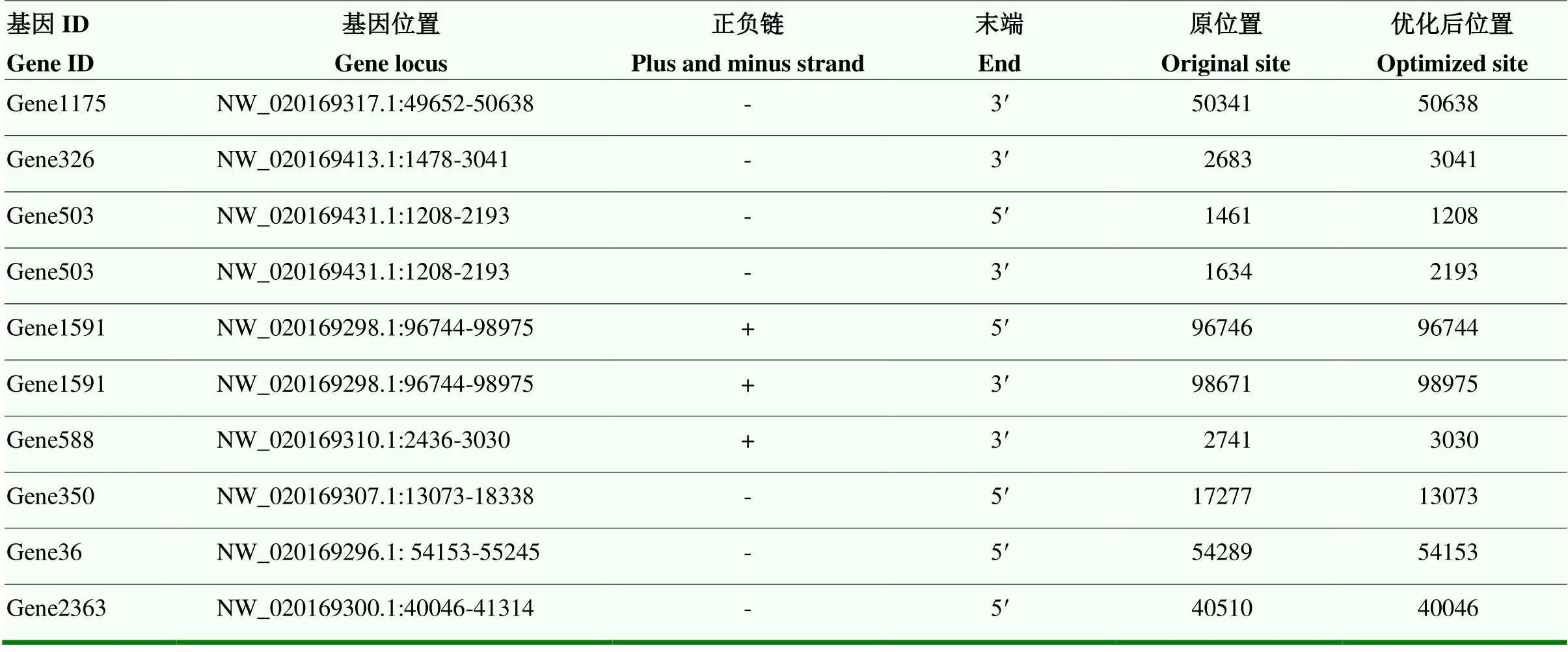
表1 东方蜜蜂微孢子虫参考基因组中10个基因的结构优化信息概要
GO数据库注释结果显示,东方蜜蜂微孢子虫的新基因还能注释到生物学进程大类的15个条目,包括细胞进程(237)、代谢进程(225)和单一组织进程(114)等;分子功能大类的10个条目,包括催化活性(219)、结合(207)和结构分子活性(10)等;细胞组分大类的10个条目,包括细胞(214)、细胞组件(212)和细胞器(158)等(图2)。此外,上述新基因还可注释到KEGG数据库的58条通路,注释基因数最多的前5位通路分别是真核生物核糖体的生物合成(24),嘧啶代谢(17),抗生素的生物合成(16),内质网的蛋白加工(16)和嘌呤代谢(15)(图3)。括号内的数字代表注释上的新基因数量。

1:细胞外区域Extracellular region;2:细胞Cell;3:细胞膜Cell membrane;4:细胞膜内腔Membrane-enclosed lumen;5:高分子复合物Macromolecular complex;6:细胞器Organelle;7:细胞器组件Organelle part;8:细胞膜组件Cell membrane part;9:细胞组件Cell part;10:超分子复合物Supramolecular complex;11:转录因子活性,蛋白质结合Transcription factor activity, protein binding;12:核酸结合转录因子活性Nucleic acid binding transcription factor activity;13:催化活性Catalytic activity;14:信号转导因子活性Signal transducer activity;15:结构分子活性Structural molecule activity;16:转运活性Transporter activity;17:结合Binding;18:电子载体活性Electron carrier activity;19:抗氧化活性Antioxidant activity;20:分子功能调节因子Molecular function regulator;21:繁殖Reproduction;22:代谢进程Metabolic process;23:细胞进程Cellular process;24:生殖进程Reproductive process;25:生物黏附Biological adhesion;26:信号Signaling;27:发育进程Developmental process;28:生长Growth;29:单一组织进程Single-organism process;30:应激反应Response to stimulus;31:定位Localization;32:多组织进程Multi-organism process;33:生物调控Biological regulation;34:细胞成分组织或生物合成Cellular component organization or biogenesis;35:解毒Detoxification
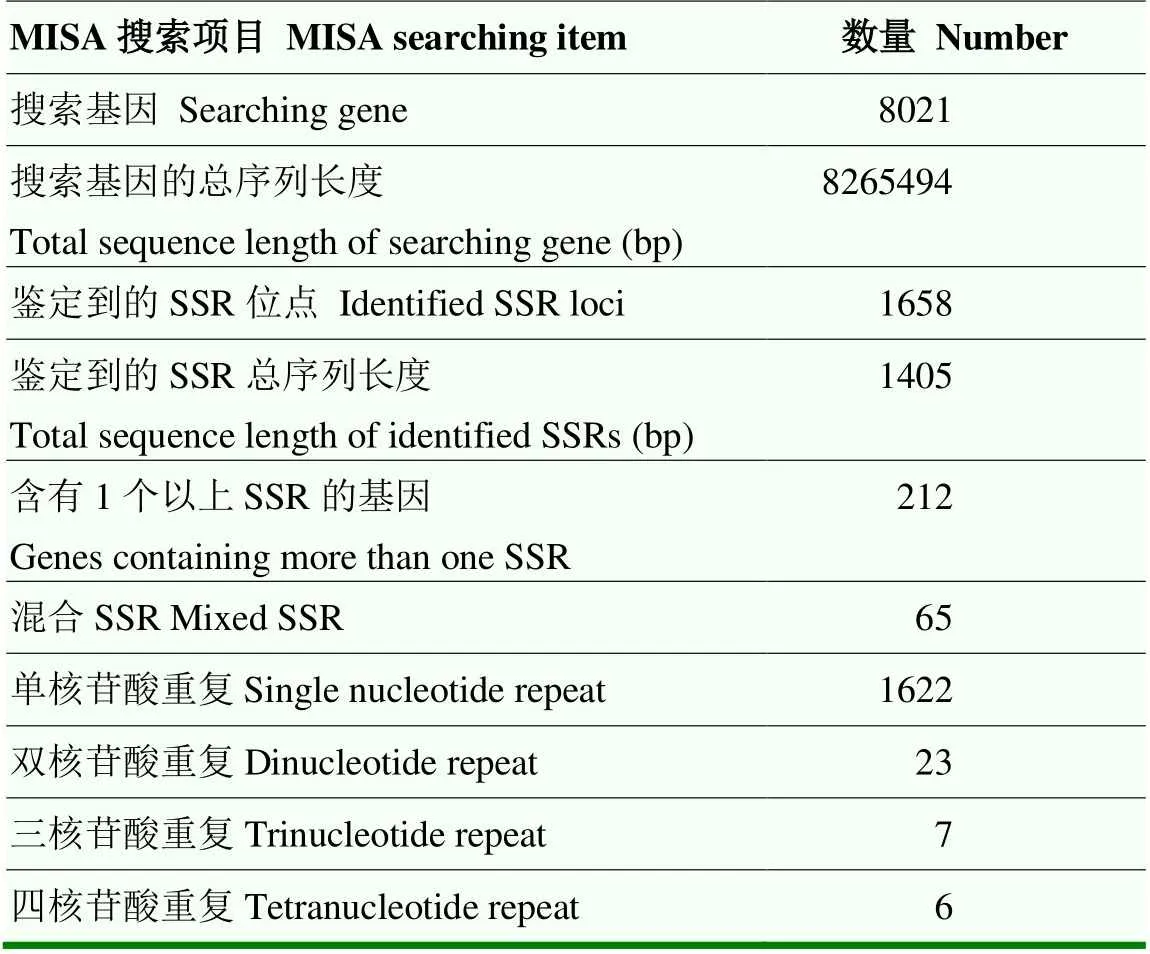
表2 基于MISA的东方蜜蜂微孢子虫SSR的搜索结果
2.4 东方蜜蜂微孢子虫的新转录本鉴定及数据库注释
共鉴定出6 164条新转录本,其中分别有6 141、2 808、2 932、3 196和2 585条新转录本可注释到Nr、KOG、eggNOG、GO和KEGG数据库。新转录本注释数量最多的物种是东方蜜蜂微孢子虫(5 512),其次为蜜蜂微孢子虫(263)和家蚕微孢子虫()(156)(图4-A)。新转录本可注释到KOG数据库的25个功能类别,注释转录本数最多的是翻译、核糖体结构和生物合成(370),其次是转录(337),翻译后修饰、蛋白折叠和分子伴侣(327),复制、重组和修复(319)及RNA的加工与修饰(281)(图4-B)。此外,新转录本可注释到eggNOG数据库的25个功能类别,注释转录本数最多的是未知功能(557),其次是翻译、核糖体结构和生物合成(433),复制、重组和修复(391),转录(320)及翻译后修饰、蛋白折叠和分子伴侣(297)(图4-C)。括号内的数字代表注释上的新转录本数量。
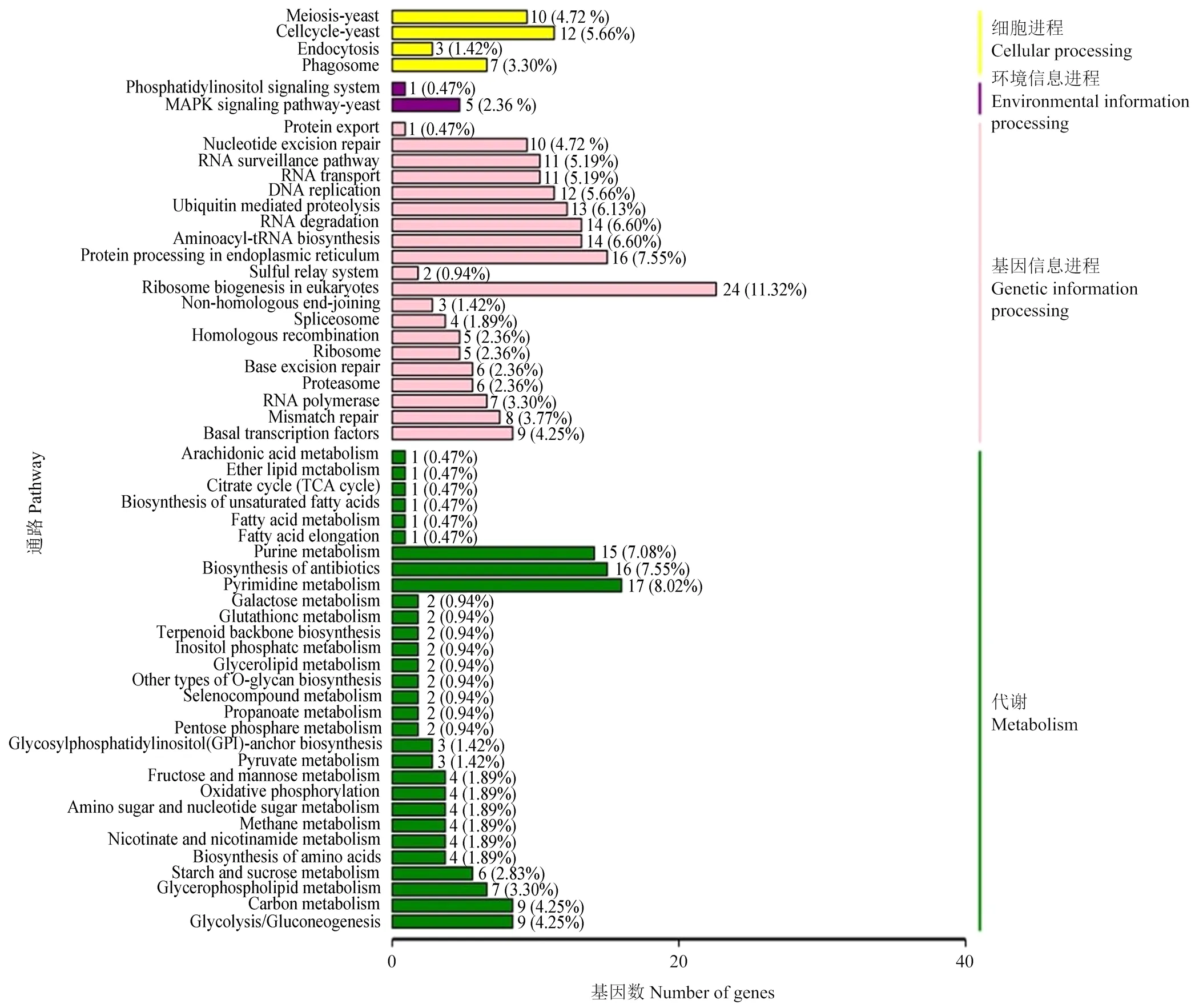
图3 东方蜜蜂微孢子虫新基因的KEGG数据库注释
GO数据库注释结果显示,东方蜜蜂微孢子虫的新转录本还能注释到生物学进程大类的16个条目,包括细胞进程(1 973)、代谢进程(1 814)和单一组织进程(856)等;分子功能大类的10个条目,包括结合(1 711)、催化活性(1 561)和结构分子活性(147)等;细胞组分大类的15个条目,包括细胞组件(1 819)、细胞(1 816)和细胞器(1 392)等(图5)。此外,上述新基因还可注释到KEGG数据库的58条通路,包括真核生物核糖体的生物合成(148)、嘧啶代谢(146)、嘌呤代谢(139)、RNA降解(116)及细胞周期-酵母(113)等(图6)。括号内数字代表注释上的新转录本数量。
2.5 东方蜜蜂微孢子虫的完整ORF预测
利用软件共预测出2 353个完整ORF,它们的长度分布介于0—400 aa,其中分布在0—100 aa的ORF数量最多,为1 697个;分布在100—200、200—300和300—400 aa的ORF分别有603、47和6个(图7)。
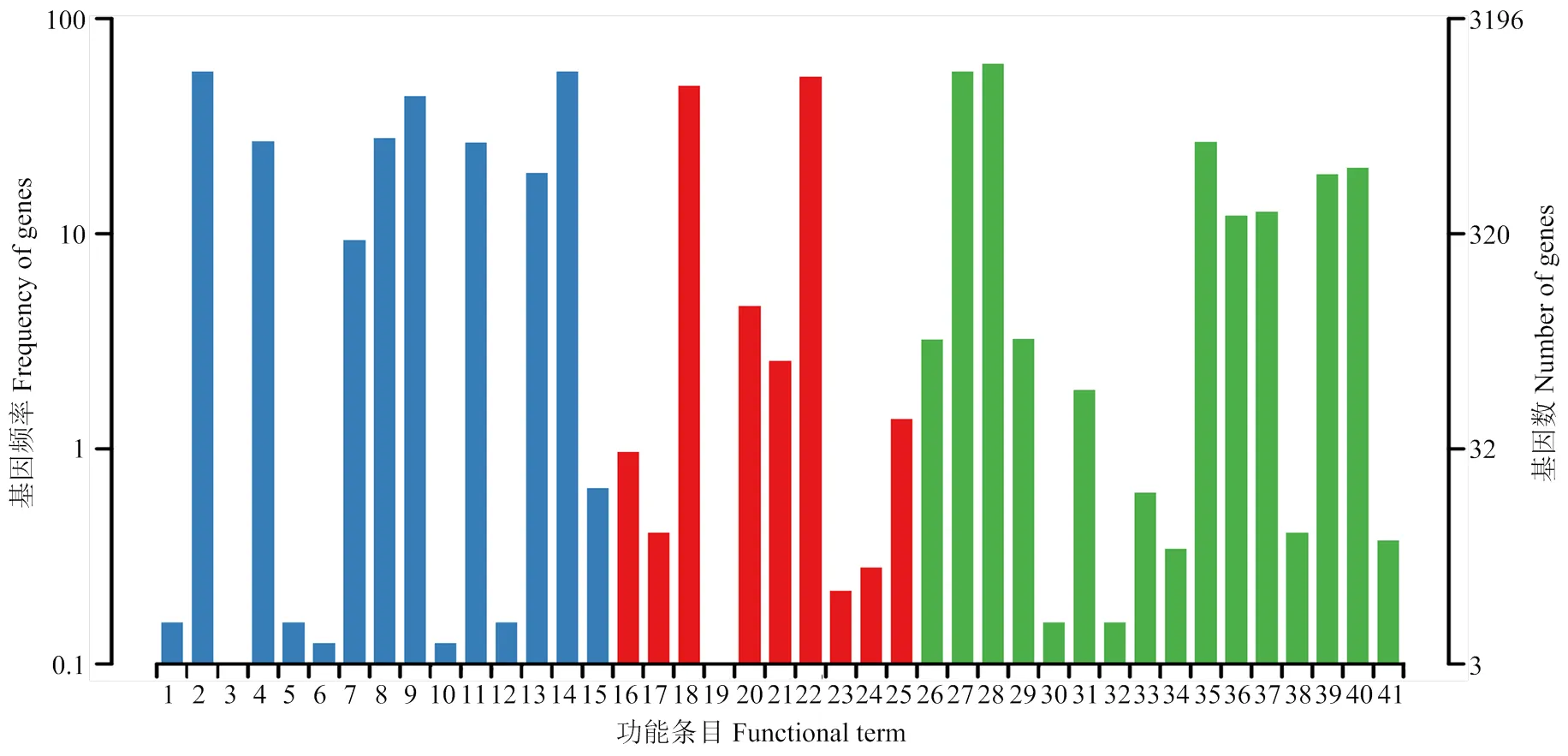
1:细胞外区域Extracellular region;2:细胞Cell;3:拟核Nucleoid;4:细胞膜Cell membrane;5:病毒Virion;6:细胞连接Cell junction;7:细胞膜内腔Membrane-enclosed lumen;8:高分子复合物Macromolecular complex;9:细胞器Organelle;10:胞外区组件Extracellular region part;11:细胞器组件Organelle part;12:病毒组件Virion part;13:细胞膜组件Cell membrane part;14:细胞组件Cell part;15:超分子复合物Supramolecular complex;16:转录因子活性,蛋白质结合Transcription factor activity, protein binding;17:核酸结合转录因子活性Nucleic acid binding transcription factor activity;18:催化活性Catalytic activity;19:信号转导因子活性Signal transducer activity;20:结构分子活性Structural molecule activity;21:转运活性Transporter activity;22:结合Binding;23:电子载体活性Electron carrier activity;24:分子功能调控器Molecular function regulator;25:抗氧化活性Antioxidant activity;26:繁殖Reproduction;27:代谢进程Metabolic process;28:细胞进程Cellular process;29:生殖进程Reproductive process;30:生物黏附Biological adhesion;31:信号Signaling;32:多组织进程Multicellular organismal process;33:发育进程Developmental process;34:生长Growth;35:单一组织进程Single-organism process;36:应激反应Response to stimulus;37:定位Localization;38:多细胞组织进程Multi-organism process;39:生物调控Biological regulation;40:细胞成分组织或生物合成Cellular component organization or biogenesis;41:解毒Detoxification
3 讨论
近十几年来,二代测序技术的迅速发展和应用有力推动了动物、植物和微生物的基因组和转录组研究,存储于公共数据库(如NCBI SRA数据库)的海量二代转录组测序数据已成为完善物种基因组序列和功能注释的宝贵资源[22-23]。相对于一代和二代测序技术,Nanopore长读段测序技术具有超长读长(平均读长可达15 kb)的显著优势,不需要对测序读段进行拼接就能获得转录本的全长序列,所测即所得[24]。本研究利用前期已获得的全长转录组数据对东方蜜蜂微孢子虫的基因组注释进行完善,预测出2 353个完整ORF,分别延长了1 182和1 158个基因的5′ UTR和3′ UTR,发掘出1 658个SSR位点,此外鉴定到954个新基因和6 164条新转录本并对它们进行了功能注释。此为利用三代转录组测序数据完善蜜蜂病原基因组注释的首例报道。需要注意的是,本研究使用的全长转录组数据来源于东方蜜蜂微孢子虫的纯净孢子,而孢子是病原的休眠态,仅维持必要的低水平代谢[25],表达的转录本必然与病原在侵染过程表达的转录本存在差异。目前,笔者团队已获得东方蜜蜂微孢子虫感染7 d和10 d的意大利蜜蜂(,简称意蜂)和中华蜜蜂(,简称中蜂)工蜂中肠的Nanopore长读段测序数据(未发表数据),下一步将从上述混合数据中筛滤出纯净的病原全长转录组数据,从而进一步对现有的参考基因组注释进行补充和完善。
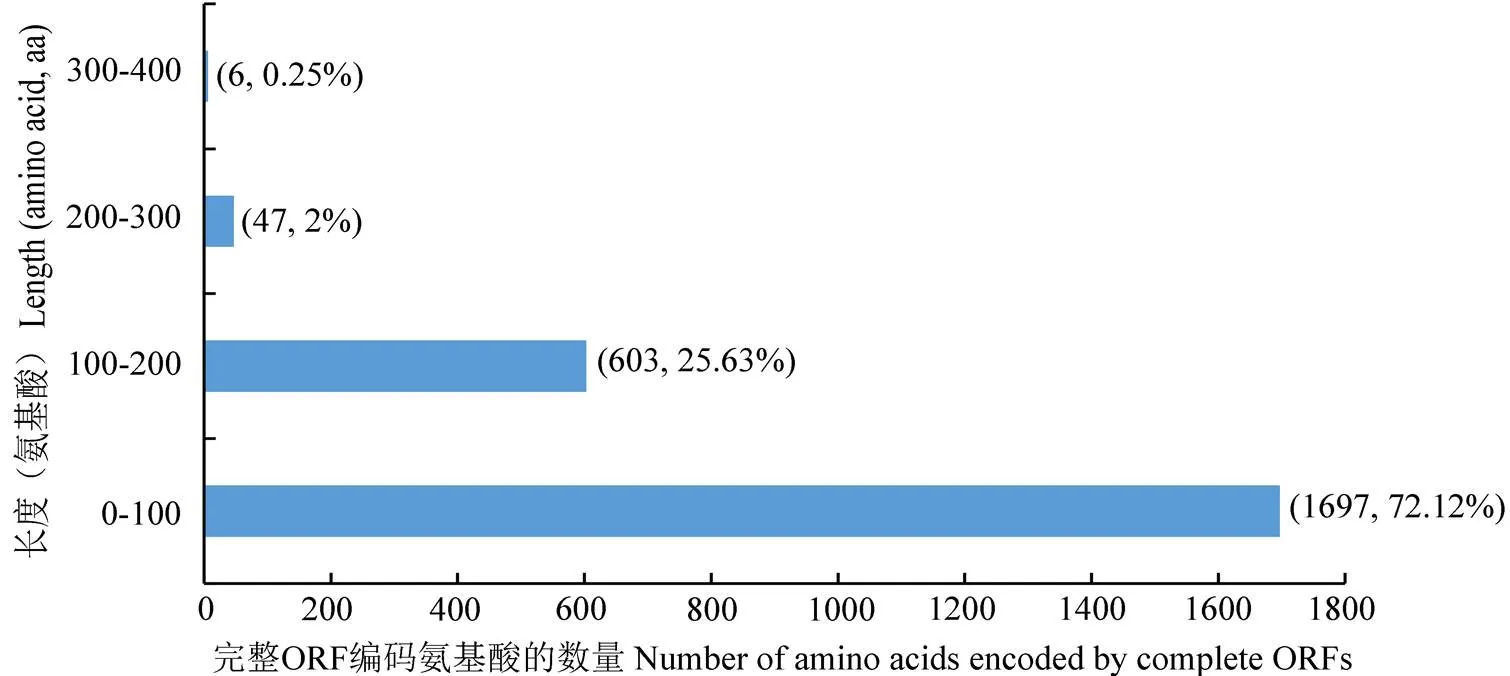
图7 东方蜜蜂微孢子虫的完整ORF编码氨基酸的长度分布
真核生物的基因表达调控与mRNA的UTR密切相关,例如mRNA的5′ UTR不仅能通过与反式作用因子结合调控翻译起始,还能通过控制mRNA的半衰期影响其稳定性;miRNA的种子序列能够与mRNA的3′ UTR靶向结合,从而抑制mRNA的翻译或使其降解[26]。前期研究中,笔者团队利用Illumina HiSeq技术对东方蜜蜂微孢子虫的纯净孢子进行测序,基于218 468 218条有效读段(clean reads)分别延长了6个已注释基因的5′ UTR和4个已注释基因的3′ UTR[27]。本研究基于东方蜜蜂微孢子虫的Nanopore全长转录组数据分别对1 182和1 158个基因的5′ UTR和3′ UTR进行了延长,说明三代测序数据较之二代测序数据可以大幅度提高已注释基因的结构优化质量,经优化的5′ UTR和3′ UTR对于深入研究东方蜜蜂微孢子虫的基因表达调控具有重要意义。
SSR是以1—6个核苷酸为重复单元组成的简单串联重复序列,作为第二代分子标记,SSR具有共显性遗传、重复性好、实验操作易及多态性高等优点[28]。SSR开发的传统方法以文库构建法为主,过程繁杂、费时费力且效率低下[29]。随着二代测序技术的不断进步和转录组数据的持续增多,人们开始利用测序得到的和公共数据库存储的二代转录组数据大规模开发SSR[30-31]。笔者团队前期也利用二代转录组数据大规模开发和验证了蜜蜂球囊菌()、中蜂和意蜂的SSR[32-34],证实了该方法的可行性。目前,东方蜜蜂微孢子虫的SSR严重缺乏。本研究基于东方蜜蜂微孢子虫的高质量全长转录组数据发掘出1 658个未注释的SSR位点,为现有的参考基因组的注释提供了有益补充。在前期研究中,利用蜜蜂球囊菌的二代转录组数据开发出7 968个SSR,其中最主要的重复类型为三核苷酸重复(53.15 %);此外,鉴定出13 448个中蜂SSR和6 312个意蜂SSR,其中最丰富的重复类型均为双核苷酸重复,占比分别达到58.03%和54.42%。本研究发现,东方蜜蜂微孢子虫的SSR中单核苷酸重复最为丰富,占比高达97.83%,与球囊菌和中蜂的研究结果存在差异,说明不同物种SSR的重复类型具有物种特异性。然而,对于沙葱萤叶甲()[35]、扶桑绵粉蚧()[31]和黄粉虫()[30]等昆虫,SSR的主要重复类型为单核苷酸重复,与本研究中东方蜜蜂微孢子虫SSR的主要重复类型一致,说明有些物种SSR的重复类型具有共性。此外,通过比较本研究鉴定到的SSR位点数与前期基于二代测序数据鉴定到的SSR位点数[32-34],发现前者的数量明显少于后者。对于哪种方法的准确性更高、假阳性更低,仍需要进一步深入研究。未来的工作重点是针对发掘出的SSR位点批量设计特异性引物,通过PCR扩增和毛细管电泳验证SSR的有效性及多态性,并将经验证的SSR应用于养蜂生产中东方蜜蜂微孢子虫的菌株鉴定、遗传分化及基因定位等研究。
现有的东方蜜蜂微孢子虫参考基因组(assembly ASM98816v1)共注释了3 264个基因,包括3 209个蛋白编码基因,35个tRNA基因,18个假基因和2个rRNA基因[8]。笔者团队前期基于东方蜜蜂微孢子虫的二代转录组数据仅鉴定出27个新基因[27]。本研究共鉴定到954个参考基因组未注释的新基因,占目前注释基因总数的约30%,说明基于Nanopore全长转录组数据能够高效挖掘新基因。本研究中,共有951个新基因可注释到Nr数据库,注释数量最多的物种是东方蜜蜂微孢子虫(798,83.91%),与实际情况相符,其次为蜜蜂微孢子虫(55),体现了二者同属属,亲缘关系近;分别有333、371、422和321个新基因可注释到KOG、eggNOG、GO和KEGG数据库,获得功能注释信息的新基因数量仍然偏少。一是由于目前还没有建立东方蜜蜂微孢子虫的转基因操作技术体系,导致绝大多数的基因功能尚未明确;二是上述4个数据库收录的东方蜜蜂微孢子虫及其近缘物种的功能注释信息还比较少,需要更多的研究数据对其进行持续补充。此外,本研究还鉴定出6 164条参考基因组未注释的新转录本,这些含有全长序列的新转录本为将来的基因克隆和功能研究提供了宝贵的数据资源。鉴定到的6 141条新转录本均能注释到Nr数据库,注释数量最多的物种仍为东方蜜蜂微孢子虫(5 512,89.76%),其次为蜜蜂微孢子虫(263,4.28%),与新基因的注释结果一致。分别有2 808、2 932、3 196和2 585条新转录本可注释到KOG、eggNOG、GO和KEGG数据库,这些功能注释信息可进一步完善现有的东方蜜蜂微孢子虫参考基因组的注释。
4 结论
利用高质量的Nanopore全长转录组数据对现有的东方蜜蜂微孢子虫参考基因组序列和功能注释进行了完善,为分子标记研究提供了大量SSR位点,补充了参考基因组的基因和转录本信息。
[1] WHEELER D A, SRINIVASAN M, EGHOLM M, SHEN Y, CHEN L, MCGUIRE A, HE W, CHEN Y J, MAKHIJANI V, ROTH G T,. The complete genome of an individual by massively parallel DNA sequencing. Nature, 2008, 452(7189): 872-876.
[2] XIA Q, ZHOU Z, LU C, CHENG D, DAI F, LI B, ZHAO P, ZHA X, CHENG T, CHAI C,. A draft sequence for the genome of the domesticated silkworm (). Science, 2004, 306(5703): 1937-1940.
[3] The Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee. Nature, 2006, 443(7114): 931-949.
[4] KOCHER S D, LI C, YANG W, TAN H, YI S V, YANG X, HOEKSTRA H E, ZHANG G, PIERCE N E, YU D W. The draft genome of a socially polymorphic halictid bee,. Genome Biology, 2013, 14(12): R142.
[5] PARK D, JUNG J W, CHOI B S, JAYAKODI M, LEE J, LIM J, YU Y, CHOI Y S, LEE M L, PARK Y, CHOI I Y, YANG T J, EDWARDS O R, NAH G, KWON H W. Uncovering the novel characteristics of Asian honey bee,, by whole genome sequencing. BMC Genomics, 2015, 16(1): 1.
[6] OLSEN J L, ROUZÉ P, VERHELST B, LIN Y C, BAYER T, COLLEN J, DATTOLO E, DE PAOLI E, DITTAMI S, MAUMUS F,. The genome of the seagrassreveals angiosperm adaptation to the sea. Nature, 2016, 530(7590): 331-335.
[7] ZHANG G Q, XU Q, BIAN C, TSAI W C, YEH C M, LIU K W, YOSHIDA K, ZHANG L S, CHANG S B, CHEN F,. TheLindlgenome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Scientific Reports, 2016, 6: 19029.
[8] PELIN A, SELMAN M,1 ARIS-BROSOU S, FARINELLI L, CORRADI N. Genome analyses suggest the presence of polyploidy and recent human-driven expansions in eight global populations of the honeybee pathogen. Environmental Microbiology, 2015, 17(11): 4443-4458.
[9] LIU B, ZHOU Y, LI K, HU X, WANG C, CAO G, XUE R, GONG C. The complete genome of cyprinid herpesvirus 2, a new strain isolated from. Virus Research, 2018, 256: 6-10.
[10] RHOADS A, AU K F. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics, 2015, 13(5): 278-289.
[11] MAGRINI V, GAO X, ROSA B A, MCGRATH S, ZHANG X, HALLSWORTH-PEPIN K, MARTIN J, HAWDON J, WILSON R K, MITREVA M. Improving eukaryotic genome annotation using single molecule mRNA sequencing.BMC Genomics, 2018, 19(1): 172.
[12] TANG Y T, GAO X, ROSA B A, ABUBUCKER S, HALLSWORTH- PEPIN K, MARTIN J, TYAGI R, HEIZER E, ZHANG X, BHONAGIRI-PALSIKAR V,. Genome of the human hookworm. Nature Genetics, 2014, 46(3): 261-269.
[13] MITREVA M, JASMER D P, ZARLENGA D S, WANG Z, ABUBUCKER S, MARTIN J, TAYLOR C M, YIN Y, FULTON L, MINX P,. The draft genome of the parasitic nematode. Nature Genetics, 2011, 43(3): 228-235.
[14] PENDLETON M, SEBRA R, PANG A W C, UMMAT A, FRANZEN O, RAUSCH T, STÜTZ A M, STEDMAN W, ANANTHARAMAN T, HASTIE A,. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nature Methods, 2015, 12(8): 780-786.
[15] SHIELDS E J, SHENG L, WEINER A K, GARCIA B A, BONASIO R. High-quality genome assemblies reveal long non-coding RNAs expressed in ant brains. Cell Reports, 2018, 23(10): 3078-3090.
[16] DACCORD N, CELTON J M, LINSMITH G, BECKER C, CHOISNE N, SCHIJLEN E, VAN DE GEEST H, BIANCO L, MICHELETTI D, VELASCO R,. High-qualityassembly of the apple genome and methylome dynamics of early fruit development. Nature Genetics, 2017, 49(7): 1099-1106.
[17] DONG L, LIU H, ZHANG J, YANG S, KONG G, CHU S C, CHEN N, WANG D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics, 2015, 16(1): 1039.
[18] CORNMAN R S, CHEN Y P, SCHATZ M C, STREET S, ZHAO Y, DESANY B, EGHOLM M, HUTCHISON S, PETTIS J S, LIPKIN W I, EVANS J D. Genomic analyses of the microsporidian, an emergent pathogen of honey bees. PLoS Pathogens, 2009, 5(6): e1000466.
[19] 陈华枝, 杜宇, 范小雪, 祝智威, 蒋海宾, 王杰, 范元婵, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 基于第三代纳米孔测序技术的东方蜜蜂微孢子虫全长转录组构建及注释. 昆虫学报, 2020, 63(12): 1461-1472.
CHEN H Z, DU Y, FAN X X, ZHU Z W, JIANG H B, WANG J, FAN Y C, XIONG C L, ZHENG Y Z, FU Z M, XU G J, CHEN D F, GUO R. Construction and annotation of the full-length transcriptome ofbased on the third-generation nanopore sequencing technology. Acta Entomologica Sinica, 2020, 63(12): 1461-1472. (in Chinese)
[20] 陈华枝, 范小雪, 范元婵, 王杰, 祝智威, 蒋海宾, 张文德, 隆琦, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 东方蜜蜂微孢子虫基因的可变剪接及可变腺苷酸化解析. 菌物学报, 2021, 40(1): 161-173.
CHEN H Z, FAN X X, FAN Y C, WANG J, ZHU Z W, JIANG H B, ZHANG W D, LONG Q, XIONG C L, ZHENG Y Z, FU Z M, XU G J, CHEN D F, GUO R. Analysis of alternative splicing and alternative polyadenylation ofgenes. Mycosystema, 2021, 40(1): 161-173. (in Chinese)
[21] THIEL T, MICHALEK W, VARSHNEY R, GRANER A. Exploiting EST databases for the development and characterization of gene- derived SSR-markers in barley (L.). Theoretical and Applied Genetics, 2003, 106(3): 411-422.
[22] 郭睿, 陈华枝, 童新宇, 熊翠玲, 郑燕珍, 付中民, 解彦玲, 王海朋, 赵红霞, 陈大福. 蜜蜂球囊菌基因结构优化及新基因鉴定. 中国农业大学学报, 2019, 24(1): 61-68.
GUO R, Chen H Z, Tong X Y, XIONG C L, ZHENG Y Z, FU Z M, XIE Y L, WANG H P, ZHAO H X, CHEN D F. Structural optimization of annotated genes and identification of novel genes in. Journal of China Agricultural University, 2019, 24(1): 61-68. (in Chinese)
[23] 熊翠玲, 王海朋, 郑燕珍, 付中民, 徐国均, 童新宇, 赵红霞, 陈大福, 郭睿. 基于中华蜜蜂幼虫肠道转录组数据对东方蜜蜂基因组的基因结构优化及新基因鉴定. 中国农业大学学报, 2019, 24(3): 86-93.
XIONG C L, WANG H P, ZHENG Y Z, FU Z M, XU G J, TONG X Y, ZHAO H X, CHEN D F, GUO R. Gene structure optimization and identification of novel genes ingenome: Based on the transcriptome data obtained from larval gut.Journal of China Agricultural University, 2019, 24(3): 86-93. (in Chinese)
[24] CHENG B, FURTADO A, HENRY R J. Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Giga Science, 2017, 6(11): 1-13.
[25] GUO R, CHEN D F, XIONG C L, HOU C S, ZHENG Y Z, FU Z M, LIANG Q, DIAO Q Y, ZHANG L, WANG H Q, HOU Z X, KUMAR D. First identification of long non-coding RNAs in fungal parasite. Apidologie, 2018, 49(5): 660-670.
[26] BARRETT L W, FLETCHER S, WILTON S D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements.Cellular and Molecular Life Sciences, 2012, 69(21): 3613-3634.
[27] 熊翠玲, 童新宇, 陈华枝, 耿四海, 庄天艺, 郑燕珍,付中民, 陈大福, 赵红霞, 郭睿. 东方蜜蜂微孢子虫的基因结构优化及新基因鉴定. 环境昆虫学报, 2019, 41(2): 373-379.
XIONG C L, TONG X Y, CHEN H Z, GENG S H, ZHUANG T Y, ZHENG Y Z, FU Z M, CHEN D F, ZHAO H X, GUO R. Optimization of gene structure and identification of novel genes in. Journal of Environmental Entomology, 2019, 41(2): 373-379. (in Chinese)
[28] JARNE P, LAGODA P J. Microsatellites, from molecules to populations and back. Trends in Ecology and Evolution, 1996, 11(10): 424-429.
[29] ZANE L, BARGELLONI L, PATARNELLO T. Strategies for microsatellite isolation: A review. Molecular ecology, 2002, 11(1): 1-16.
[30] 朱家颖, 吴国星, 杨斌. 基于转录组数据高通量发掘黄粉甲微卫星引物. 昆虫学报, 2013, 56(7): 724-728.
ZHU J Y, WU G X, YANG B. High-throughput discovery of SSR genetic markers in the yellow mealworm beetle,(Coleoptera: Tenebrionidae), from its transcriptome database. Acta Entomologica Sinica, 2013, 56(7): 724-728. (in Chinese)
[31] 罗梅, 张鹤, 宾淑英, 林进添. 基于转录组数据高通量发掘扶桑绵粉蚧微卫星引物. 昆虫学报, 2014, 57(4): 395-400.
LUO M, ZHANG H, BIN S Y, LIN J T. High-throughput discovery of SSR genetic markers in the mealybug,(Hemiptera: Pseudococcidae), from its transcriptome database. Acta Entomologica Sinica, 2014, 57(4): 395-400. (in Chinese)
[32] 李汶东, 熊翠玲, 王鸿权, 侯志贤, 童新宇, 张璐, 付中民, 郑燕珍, 陈大福, 郭睿. 基于RNA-seq数据大规模挖掘蜜蜂球囊菌的SSR分子标记. 福建农林大学学报(自然科学版), 2017, 46(4): 434-438.
LI W D, XIONG C L, WANG H Q, HOU Z X, TONG X Y, ZHANG L, FU Z M, ZHENG Y Z, CHEN D F, GUO R. Large scale development of SSR molecular markers ofbased on RNA-seq data. Journal of Fujian Agriculture and Forestry University (Natural Science Edition), 2017, 46(4): 434-438. (in Chinese)
[33] 熊翠玲, 张璐, 付中民, 王鸿权, 侯志贤, 童新宇, 李汶东, 郑燕珍, 陈大福, 郭睿. 基于RNA-seq数据大规模开发中华蜜蜂幼虫的SSR分子标记. 环境昆虫学报, 2017, 39(1): 68-74.
XIONG C L, ZHANG L, FU Z M, WANG H Q, HOU Z X, TONG X Y, LI W D, ZHENG Y Z, CHEN D F, GUO R. Large-scale development of SSR primers forlarvae based on its RNA-seq datasets. Journal of Environmental Entomology, 2017, 39(1): 68-74. (in Chinese)
[34] 郭睿, 陈华枝, 庄天艺, 熊翠玲, 郑燕珍, 付中民, 陈恒, 陈大福. 利用转录组数据开发意大利蜜蜂的SSR分子标记. 安徽农业大学学报, 2018, 45(3): 404-408.
GUO R, CHEN H Z, ZHUANG T Y, XIONG C L, ZHENG Y Z, FU Z M, CHEN H, CHEN D F. Exploitation of SSR markers forligustica based on transcriptome data. Journal of Anhui Agricultural University, 2018, 45(3): 404-408. (in Chinese)
[35] 张鹏飞, 周晓榕, 庞保平, 谭瑶, 常静, 高利军. 基于转录组数据高通量发掘沙葱萤叶甲微卫星引物. 应用昆虫学报, 2016, 53(5): 1058-1064.
ZHANG P F, ZHOU X R, PANG B P, TAN Y, CHANG J, GAO L J. High-throughput discovery of microsatellite markers in(Coleoptera: Chrysomelidae) from a transcriptome database. Chinese Journal of Applied Entomology, 2016, 53(5): 1058-1064. (in Chinese)
Improvement ofGenome annotation based on Nanopore full-length transcriptome data
CHEN Huazhi1, FAN Yuanchan1, JIANG Haibin1, WANG Jie1, FAN Xiaoxue1, ZHU Zhiwei1, LONG Qi1, CAI Zongbing1, ZHENG Yanzhen1, FU Zhongmin1,2, XU Guojun1, CHEN Dafu1,2, GUO Rui1,2
1College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002;2Apitherapy Research Institute, Fujian Agriculture and Forestry University, Fuzhou 350002
【】The objective of this study is to improve gene sequence and functional annotation of current reference genome ofusing previously obtained Nanopore full-length transcriptome dataset.【】TransDecoder software was used to predict open reading frames (ORFs) ofand corresponding amid acids. Comparison between full-length transcripts and transcripts annotated in reference genome was performed using gffcompare software to extend upstream sequences or downstream sequences of annotated genes’ untranslated regions and correct genes’ boundaries. MISA software was used to explore simple sequence repeat (SSR) loci within transcripts with a length above 500 bp, including single nucleotide repeat, dinucleotide repeat, trinucleotide repeat, tetranucleotide repeat, pentanucleotide repeat, hexanucleotide repeat and mixed SSR. By using Blast tool, novel genes and novel transcripts were aligned to Nr, KOG, eggnog, GO and KEGG databases to gain functional annotations.【】A total of 2 353 complete ORFs were predicted, and those ORFs with a length distribution among 0-100 aa were the predominant, reaching a ratio of 72.12% among total ORFs. Additionally, structures of 2 340genes were optimized; 5′ ends of 1 182 genes and 3′ ends of 1 158 genes were respectively prolonged. Moreover, 1 658 SSRs were identified, and the numbers of single nucleotide repeat, dinucleotide repeat, trinucleotide repeat, tetranucleotide repeat were 1 622, 23, seven and six, respectively. the density of single nucleotide repeat was the highest (182.32/Mb), followed by those of mixed SSR, dinucleotide repeat and trinucleotide repeat, reaching 6.90, 2.78 and 0.73/Mb, respectively. Further, 954 novel genes were identified, among them 951, 333, 371, 422 and 321 were respectively annotated to Nr, KOG, eggNOG, GO and KEGG databases. In addition, 6 164 novel transcripts were identified, among them 6 141, 2 808, 2 932, 3 196 and 2 585 were annotated to the aforementioned five databases, respectively. The species annotated by the highest number of new gene and new transcript wasfollowed by. 【】Our results well improve sequences and functional annotations of annotated genes in current reference genome of, and supplement and annotate a number of unannotated novel genes and transcripts. lots of SSR sites were provided for research on molecular markers, information of genes and transcripts on reference genome were supplemented.
nanopore sequencing; full-length transcript; transcriptome; genome; honeybee;

10.3864/j.issn.0578-1752.2021.06.018
2020-05-06;
2020-05-28
国家现代农业产业技术体系建设专项(CARS-44-KXJ7)、福建省自然科学基金(2018J05042)、福建农林大学杰出青年科研人才计划(xjq201814)、福建农林大学科技创新专项基金(CXZX2017342,CXZX2017343)、福建农林大学优秀硕士学位论文资助基金(陈华枝)
陈华枝,E-mail:CHZ0720@outlook.com。范元婵,E-mail:fanyc19980201@126.com。陈华枝和范元婵为同等贡献作者。通信作者郭睿,E-mail:ruiguo@fafu.edu.cn
(责任编辑 岳梅)
猜你喜欢
动物医学进展(2022年6期)2022-11-26
军事文摘(2022年16期)2022-08-24
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
四川蚕业(2022年1期)2022-06-06
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
科学大众(中学)(2015年9期)2015-10-12
