
烯烃与二氧化碳直接合成环碳酸酯的研究进展
2021-03-19 12:01:58陈静雯鲍宗必杨启炜杨亦文张治国
高校化学工程学报 2021年1期
易 敏, 陈静雯, 童 杨, 鲍宗必, 杨启炜, 杨亦文, 张治国
烯烃与二氧化碳直接合成环碳酸酯的研究进展
易 敏1,2, 陈静雯1,2, 童 杨3, 鲍宗必1,2, 杨启炜1,2, 杨亦文1,2, 张治国1,2
(1. 生物质化工教育部重点实验室, 浙江大学 化学工程与生物工程学院, 浙江 杭州 310027;2. 浙江大学衢州研究院, 浙江 衢州 324000; 3. 科技部高技术研究发展中心, 北京 100044)
二氧化碳(CO2)是一种来源丰富、价廉易得的C1资源,将二氧化碳化学固定为环碳酸酯是最具工业应用前景的CO2资源化利用途径之一。相比于环氧化物与CO2反应制备环碳酸酯,以毒性小、价格低廉的烯烃为原料,通过环氧化、CO2环加成反应一步制备环碳酸酯,由于反应路线简洁、原子经济性高,具有重要的工业应用价值。在此回顾了近年来烯烃与CO2直接合成环碳酸酯的研究进展,着重介绍了不同种类的催化剂,主要包括离子液体、金属氧化物或盐、金属有机配合物、金属有机框架材料等催化剂,并对其未来发展方向进行了展望。
二氧化碳;烯烃;一步法合成;环碳酸酯
1 前 言
二氧化碳(CO2)是主要的温室气体,也是重要的C1资源,将CO2转化为高附加值化学化工产品不仅能缓解环境污染,同时也实现对资源的有效开发,是科学界的重大战略课题[1-4]。在众多CO2资源化利用途径中,CO2与环氧化物的环加成反应由于其高原子经济性与实际应用价值而备受关注,但反应原料环氧化物制备成本较高、毒性大、不易储存,导致实际生产的经济效益偏低。若从其上游原料烯烃出发,与CO2通过环氧化-环加成串联反应直接一步合成环碳酸酯,可极大提升反应经济性与环境友好性,对传统工艺革新具有重大意义。从反应机理来看,烯烃转化为环碳酸酯的反应必须历经双键的断裂及CO2的插入2个步骤,烯烃反应产生的环氧化物或者卤醇中间体,被CO2插入得到环碳酸酯[5-7]。其中,以卤醇为反应中间体的转化路线一般需要当量或过量的卤化试剂,其生成环状碳酸酯同时,伴随大量低价值含卤副产物产生,反应过程设备要求高且易造成环境污染;相比之下,以环氧化物为中间体的反应路线更加经济环保。以烯烃为原料,经环氧化-环加成反应一步合成环碳酸酯具有原料低毒、价廉、反应路线简洁、原子经济性高等优点,实现该过程的关键在于具备氧化反应催化位点和环加成反应催化位点的双催化位点高效催化体系的开发。这一课题可追溯到1962年,早年只有少量相关专利[8-9],但近年来这一合成路线因其潜在的工业应用价值而受到广泛关注[10]。本文较系统地综述了国内外烯烃与二氧化碳直接合成环碳酸酯的催化体系研究进展。
2 离子液体催化剂
离子液体(ionic liquids,ILs)具有较好的化学稳定性、热稳定性和可调控性,以及良好的溶解性[11]。近几十年来,离子液体作为新型的绿色溶剂和特殊催化剂被广泛应用于分离和催化领域。其中,离子液体用于催化CO2与环氧化物合成环碳酸酯的研究已经非常成熟[12-15],比如季铵盐、吡啶盐、咪唑盐等对该反应表现出高催化活性。近年来,研究者们尝试将其用于催化烯烃与CO2一步合成环碳酸酯的反应。
Sun等[16]于2004年首次利用四丁基溴化铵(tetrabutylammonium bromide,TBAB)作为催化剂,以叔丁基过氧化氢(tert-butyl hydrogen peroxide,TBHP)为氧化剂,在CO2压力为1 MPa、温度为80 ℃的反应条件下成功实现了烯烃与CO2直接反应合成环碳酸酯,得到苯乙烯环碳酸酯,产率为38%。但当以H2O2或O2为氧化剂时,几乎没有目标产物生成。另外,Sun等[17]还研究发现,季铵盐阴离子的亲核性越强,其催化活性越高,因为四丁基碘化铵(tetrabutylammonium iodide,TBAI)具有还原性,在氧化剂作用下,TBAI被氧化而使活性降低,因此TBAB的催化活性最高。随后Zalomaeva等[18]于2013年采用四己基溴化铵、四庚基溴化铵催化该反应,苯乙烯环碳酸酯的产率分别为20%、17%。综合以上报道,可以得出结论:随着季铵盐的碳链长度的增加,该反应的催化效果逐渐下降。
离子液体可设计性较强、结构可调,通过引入特定的官能团如羟基、磺酸基、羧基等形成功能化离子液体,其在某些特定反应中,表现出较传统离子液体更加优异的催化效果。2019年,Liu等[19]报道了一种双功能离子液体催化剂,在无溶剂的条件下,[CCIm][HCO3]可以与CC-CO2化合物相互转化(图1),HCO3−和CC-CO2分别催化烯烃环氧化和环氧化物环加成反应。实验结果表明,在以TBHP为氧化剂,CO2压力为2 MPa,在65 ℃、反应30 h的条件下,一系列芳香族烯烃能成功转化为相应的环碳酸酯,其中,对甲基苯乙烯转化率为93%,产品选择性可达85%。这种通过催化中心内部的相互转化构建多催化位点的策略,为多功能催化剂的构建提供了新的思路。
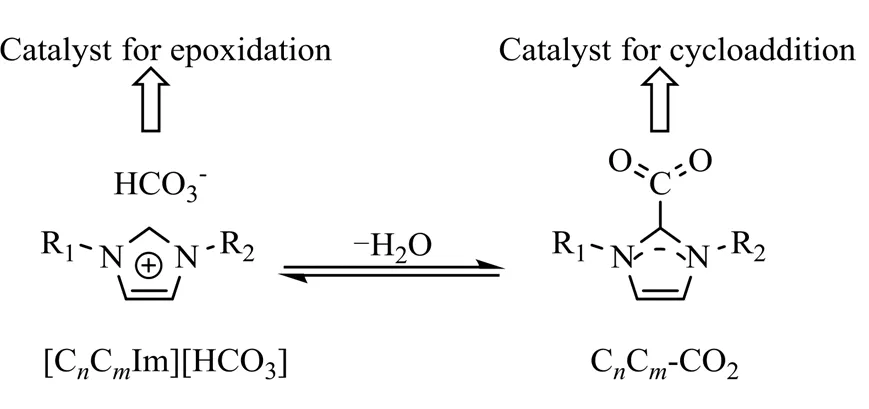
图1 [CnCmIm][HCO3]与CnCm-CO2的相互转化[19]
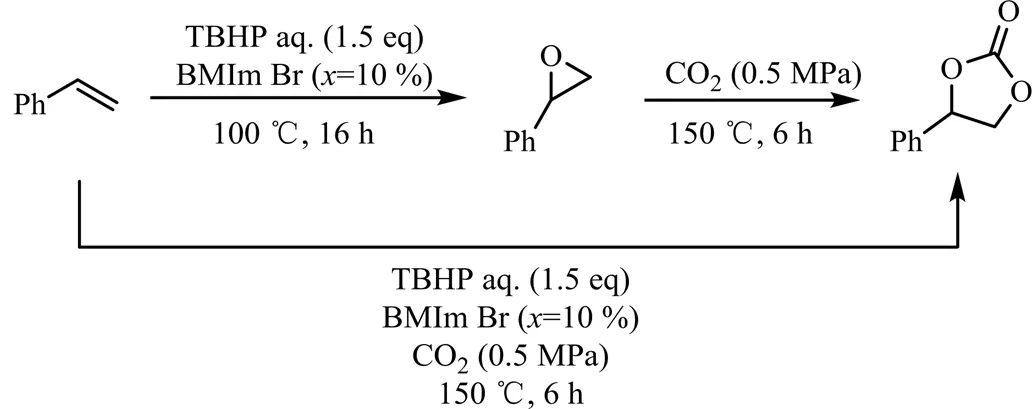
图2 BMIm Br催化苯乙烯两步或一步反应合成环碳酸酯[20]
随着季铵盐被报道用于催化烯烃与CO2一步合成环碳酸酯,Girard等[20]采用咪唑溴盐BMIm Br催化苯乙烯与CO2一步反应合成环碳酸酯(见图2)。结果表明,以TBHP为氧化剂,通过两段控温,在100 ℃下反应16 h,再升温至150 ℃,同时通入0.5 MPa CO2继续反应6 h,环碳酸酯产率接近63%。若直接一步反应,环碳酸酯的产率仅为36%。
3 金属氧化物或金属盐催化剂
金属氧化物和金属盐都是比较常见的催化剂。2000年,Aresta等[21]报道了一系列氧化物(MgO、Ag2O、Fe2O3、Nb2O5、MoO3、Ta2O5、La2O3、V2O5、ZnO等)作为催化剂用于苯乙烯与CO2反应合成环碳酸酯,在O2压力为0.5 MPa,CO2压力为4.5 MPa,反应温度为110 ℃,以DMF为溶剂的条件下反应5 h,实验结果表明苯乙烯环碳酸酯产率较低,且有较多的副产物(苯甲醛、苯乙醛、苯乙酮等)产生,其中Nb2O5催化效果相对最佳,苯乙烯转化率为27%,苯乙烯环碳酸酯的产率接近17%。
Wang等[22]将一系列钨酸盐(Na2H5P(W2O7)6、Na2WO4、Cs2.5H0.5PW12O40和(NH4)3PW12O4)与TBAB组成二元催化体系用于催化该反应(见图3 (a))。在该催化体系中,与生成环氧化物中间体不同,苯乙烯被钨酸盐催化氧化成邻溴醇中间体,邻溴醇中间体在碱作用下与CO2反应生成苯乙烯环碳酸酯,其中以Na2H5P(W2O7)6为催化剂时,苯乙烯环碳酸酯的产率为68%,然而该体系中总是伴随副产物苯甲酸苯甲酰甲酯的生成,并且在无碱存在条件下,反应优先生成该副产物(见图3 (b))。随后Xie等[23]又报道了另一无机盐K2S2O8与NaBr组成的二元催化体系促进苯乙烯到环碳酸酯的转化,反应机理类似,苯乙烯环碳酸酯的产率可提升至79%。
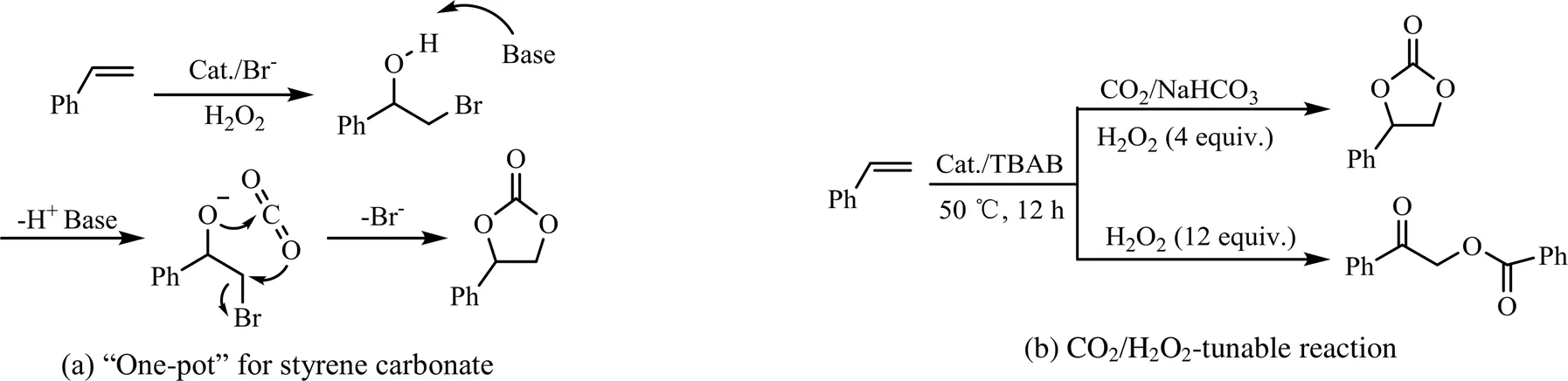
图3 Na2H5P(W2O7)6/TBAB催化苯乙烯与CO2直接合成环碳酸酯[22]
部分金属氧化物表面能够形成具备活化CO2作用的“受阻”Lewis酸碱对(frustrated Lewis pairs, FLPs),基于金属氧化物在催化氧化领域广泛应用的基础,2019年,Zhang等[24]在CeO2(110)表面构建固体FLPs位点,用于催化烯烃与CO2合成环碳酸酯的串联反应。实验结果表明,在相同实验条件下,与无FPLs位点的NO-CeO2和NC-CeO2催化剂相比,具有FLPs位点的NR-CeO2作为催化剂时,反应生成的副产物(二醇类化合物)极少,催化活性及对环碳酸酯的选择性最高(苯乙烯转化率94%,环碳酸酯选择性94%)。研究表明,在该催化体系中,原位生成的环氧化物与Ce3+具有强相互作用,从而阻碍了环氧化物的释放,并导致环氧化物在酸性Ce3+作用下进一步水解成二醇,而具有FLPs位点的NR-CeO2可以减弱这种相互作用,使原位生成的环氧化物从催化剂中释放,抑制了其水解,从而达到提高环碳酸酯产率的目的。
4 金属有机配合物催化剂
金属有机配合物常用的配体种类繁多,相应地,金属有机配合物多种多样。随着金属有机化学的飞速发展以及学者们对环氧化物与CO2反应本质的认知,很多金属配位化合物被广泛应用于环氧化物与CO2环加成反应[25-29]。此外,不少过渡金属如锰、钴、铬等能高效催化氧化反应的发生[30-33],因此通过设计并开发过渡金属配位化合物有望实现烯烃氧化羧化反应合成环碳酸酯。目前不少金属配合物如乙酰丙酮配位金属化合物、金属卟啉配合物、金属Salen配合物等被报道能有效催化烯烃转化为环碳酸酯。
Chen等[6]将钼金属配位化合物MoO2(acac)2(acetylacetonate,acac)协同季铵盐TBAB用于烯烃与CO2反应合成环碳酸酯的反应体系中(见图4)。在该反应中,MoO2(acac)2作为环氧化反应的催化中心,催化烯烃环氧化,继而环氧化物中间体与原位加入的TBAB和CO2作用转化为相应的环碳酸酯,其中,环己烯碳酸酯产率最高(84%)。
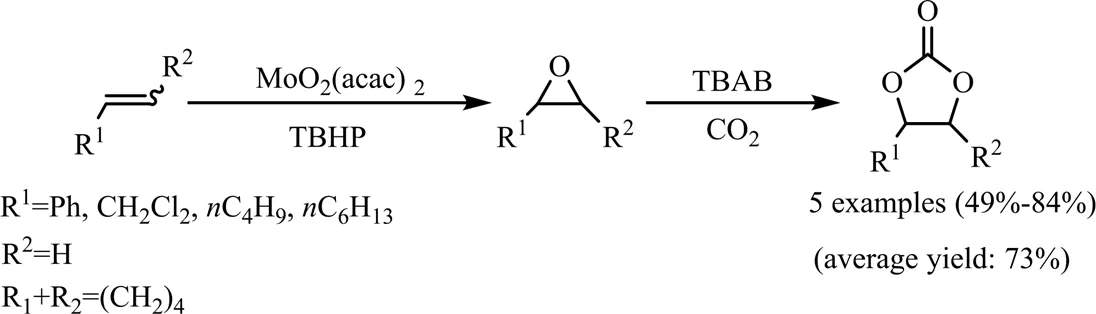
图4 MoO2(acac)2/TBAB催化烯烃与CO2反应合成环碳酸酯[6]
烯烃经环氧化-环加成合成环碳酸酯的反应一般涉及多个催化活性中心,均相催化体系成分较为复杂,给产品分离与催化剂回收带来诸多不便,因此,均相催化剂的非均相化具有重大的意义。Kumar等[34]将乙酰丙酮钴Co(acac)2和三苯基溴化磷固定在壳聚糖包覆的磁性纳米颗粒的表面,构建了双功能催化剂Co(acac)2-QPB@MCS(见图5),在分子氧为氧化剂,异丁醛为还原剂的条件下,实现了烯烃一锅氧化羧化反应合成环碳酸酯。反应结束后,可以通过外部磁铁回收磁性催化剂,该催化剂重复使用性能优良。

图5 Co(acac)2-QPB@MCS催化烯烃直接转化为环碳酸酯[34]
金属卟啉具有大环共轭芳香结构,与自然界中酶有相似的活性中心,是高效仿生均相催化剂,在催化领域备受关注[35-37]。Bai等[38]开发了在O2-CO2混合气中将烯烃转化为环碳酸酯的方法。在季铵盐TBAX (X=F−,Cl−,Br−,I−,Br3−)为助催化剂的条件下,钌卟啉RuTPP(O)2对烯烃氧化-羧化反应表现出良好的催化活性,优化条件下苯乙烯环碳酸酯产率达76%。同时,该课题组提出了可能的反应机理(见图6):RuTPP(O)2参与苯乙烯环氧化的同时,又为环氧化物中间体与CO2的环加成反应提供了Lewis酸位点。
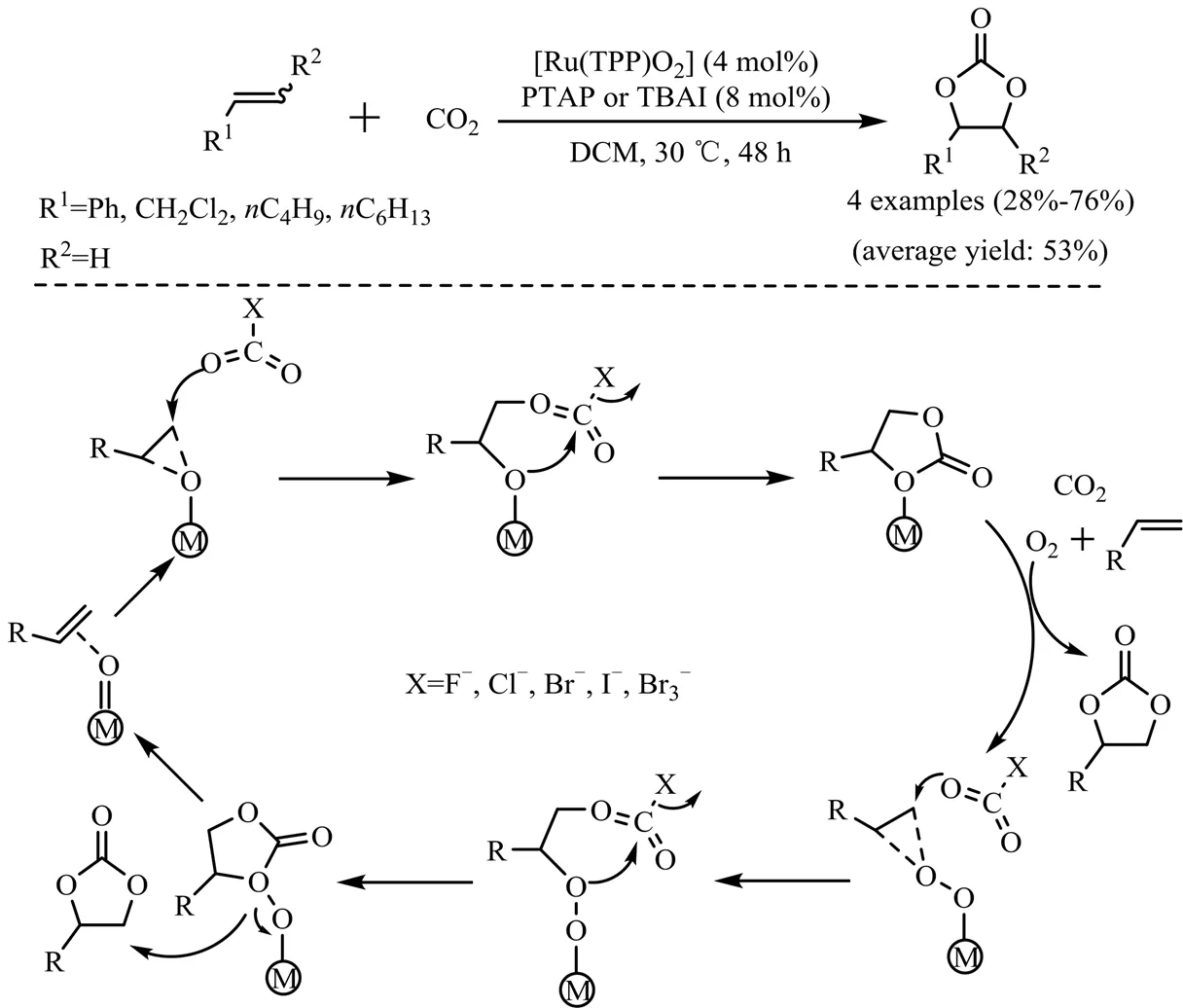
图6 Ru(TPP)O2/TBAX协同催化烯烃-O2-CO2反应合成环碳酸酯机理[38]
Dias等[39]根据烯烃转化成环碳酸酯历经两步反应的实质,将2种分别具有催化环氧化反应和环加成反应活性的金属卟啉作为催化剂,在两步投料和分段控温的条件下,MnOAc-TDCPP/CrCl-TPPCF3组成的催化体系具有良好的催化活性,其中苯乙烯转化率高达99%,苯乙烯环碳酸酯选择性为70%。为改善催化剂的循环使用性能,该研究团队还将均相金属卟啉分别固定于磁性纳米粒子上(见图7),在非均相双催化剂MNP@SiO2-8Mn/MNP@SiO2-4Cr作用下,苯乙烯环碳酸酯的产率可达40% 以上,催化剂经3次循环利用仍能保持催化活性,环碳酸酯的选择性略下降。
金属Salen配合物共轭性强,且结构具有可设计性,是一类重要的有机反应催化剂[40-42]。Qiu等[43]于2020年将均相的Salen(Pd)固定到FeNi3/DFNS纳米颗粒上(见图8),构建FeNi3/DFNS/salen/Pd(II)非均相催化剂。将其用于催化烯烃与CO2一步合成环碳酸酯,在苯胺存在的条件下,分别以O2、H2O2、单过硫酸氢钾(Oxone)、TBHP为氧化剂,结果表明以TBHP为氧化剂时,环碳酸酯的产率最高,且能催化一系列取代苯乙烯高效转化成相应的环碳酸酯(产率>89%)。
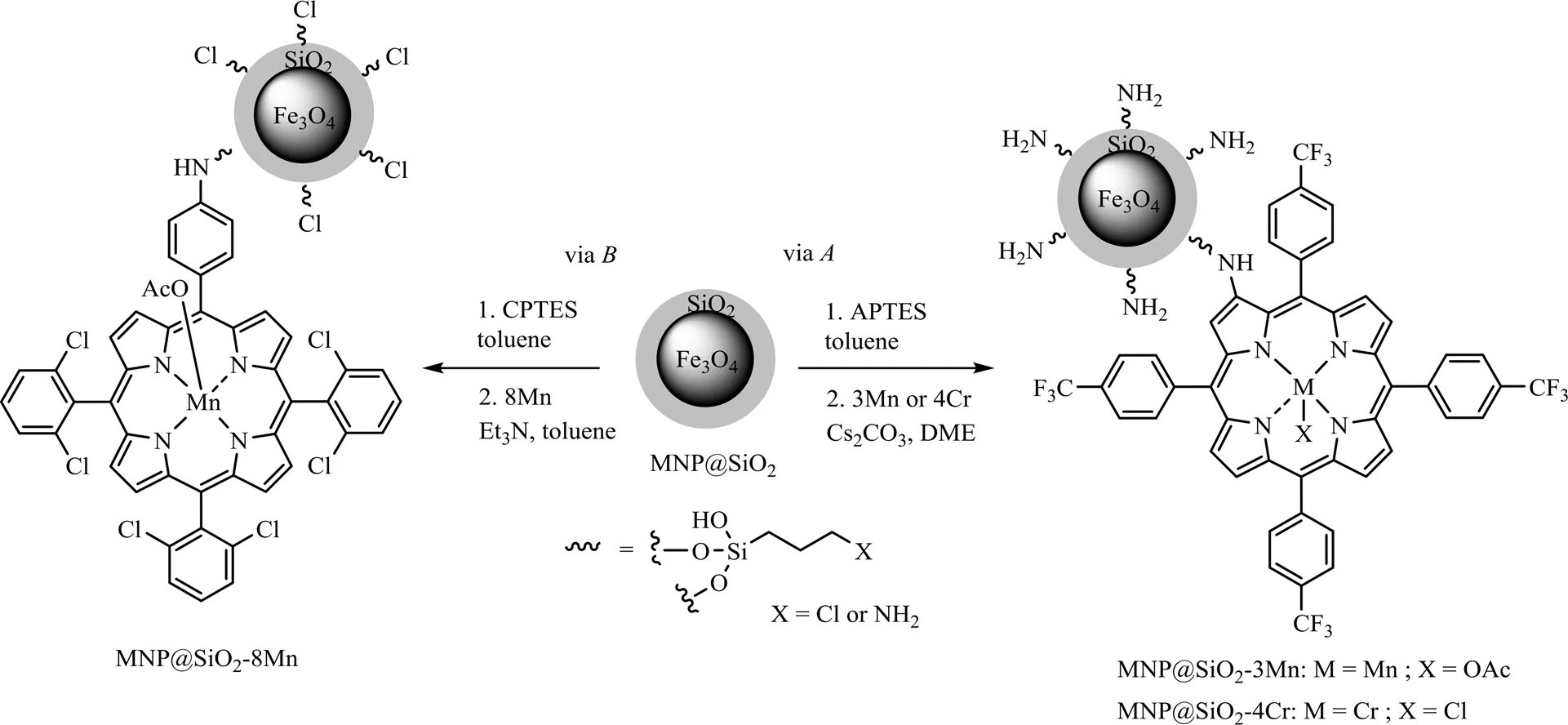
图7 杂化金属卟啉磁性材料的制备[39]
5 金属有机框架材料催化剂
金属-有机框架材料(metal-organic frameworks,MOFs)是一种由无机金属节点和有桥联配体组成的新型多孔杂化材料,其拓扑结构丰富多样且可设计性很强,被广泛应用于各种非均相催化反应中。其中,MOFs应用于催化环氧化物与CO2环加成反应合成环碳酸酯的工作被广泛报道[44-48],此外近年来不少研究表明MOFs在较低温的反应条件下也可以促进烯烃环氧化反应的发生[49-52]。因此,化学家们尝试将MOFs用于催化烯烃与CO2一步合成环碳酸酯。Zalomaeva等[18]利用性能稳定的Cr-MIL-101为催化剂、TBAB为助催化剂催化苯乙烯氧化羧化合成环碳酸酯,该研究以TBHP为氧化剂,虽然反应条件温和,但苯乙烯转化率为17%,环碳酸酯的选择性仅为25%,且反应中有大量的苯甲醛(选择性为69%)生成;然而,Cr-MIL-101/TBAB可以高效催化环氧化物与CO2的环加成反应,能使98%的氧化苯乙烯转化,苯乙烯环碳酸酯的产率高达95%。
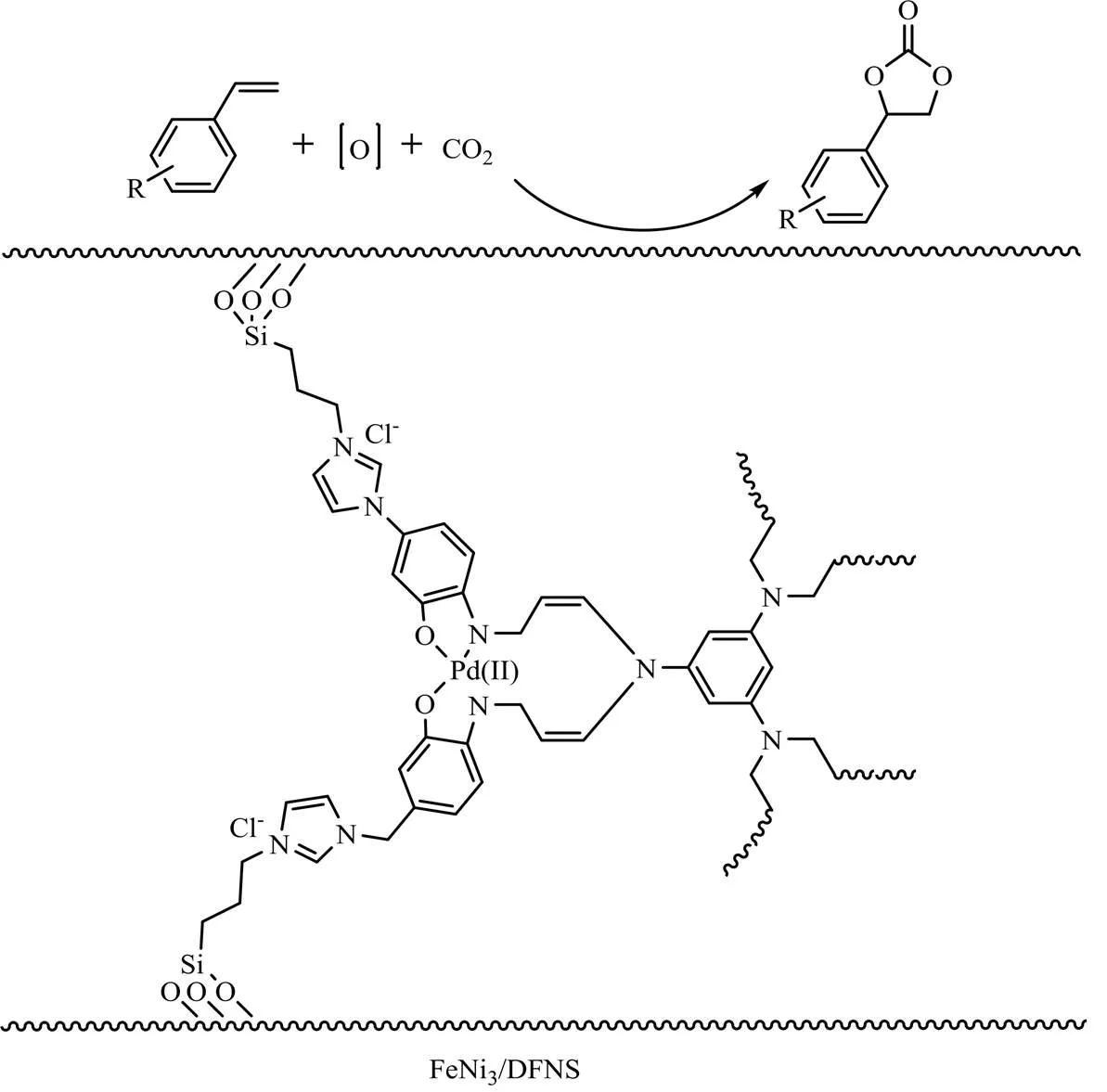
图8 FeNi3/DFNS/salen/Pd(II)催化取代苯乙烯转化为环碳酸酯[43]
除金属节点外,MOFs的功能化配体、负载的金属纳米颗粒都可以作为催化位点,MOFs的多功能化对连续催化多步反应具有重要意义。Han等[5]报道了一种具备多催化位点的MOF材料,该小组将具有良好催化环氧化功能的keggin型多金属氧酸盐(polyoxometalates,POMs)阴离子[ZnW12O40]5−引入MOF中(见图9),使所构筑的材料具有催化环氧化能力。此外组成MOF的手性吡咯配体、氨基配体以及配位不饱和金属中心,使所得的POM@MOF催化剂具备Lewis酸位点以及活化CO2的性能。实验结果表明,POM@MOF适用于多种烯烃到相应环碳酸酯的催化转化,且其中苯乙烯环碳酸酯的产率最高(92%)。

图9 POM@MOF催化苯乙烯氧化-羧化合成环碳酸酯[5]
同样地,Han等[53]于2018年采用具有催化氧化功能的钼氧化物、铜以及联吡啶(bipyridine,BPY)合成了具有Cu3(μ3-OH)2二级结构单元的双功能MOF催化剂CuMo-BPY(见图10)。实验结果表明,在以TBHP为氧化剂,TBAB为助催化剂的条件下,CuMo-BPY能有效催化烯烃环氧化-环加成反应合成环碳酸酯,其中苯乙烯环碳酸酯产率为55%。在该反应体系中,钼金属中心被TBHP氧化而转化为过氧化钼中间体,进而亲电进攻烯烃双键,生成环氧化物中间体,碱性的Cu3(μ3-OH)2结构单元使CO2活化,同时溴阴离子对环氧化物中间体的β碳原子亲核进攻,使环氧化物开环,进而与活化的CO2反应闭环生成环碳酸酯。
2020年,Ke等[54]为可控的多功能催化剂的构建提供了一种新策略。具体地,他们通过相互静电作用将咪唑型离子液体和Au纳米颗粒逐步固定在MOF材料的配体磺酸基上,构建了具有高度有序催化活性位点的多功能催化剂Au@[IM+]/[MIL-101-SO3−]。在烯烃环氧化-环加成的反应过程中,Au纳米颗粒和咪唑阳离子分别催化环氧化与环加成反应,各催化活性位点之间的强静电吸引力与高度有序排列使催化位点紧密接触,有利于串联反应的进行;此外,MOF催化剂的孔道使底物分子富集,为串联反应提供了密闭的催化空间。结果表明,Au@[IM+]/[MIL-101-SO3−]能够有效催化烯烃到环碳酸酯的转化,苯乙烯环碳酸酯产率可达74%以上,且该催化剂稳定性良好(可重复利用8次以上)。
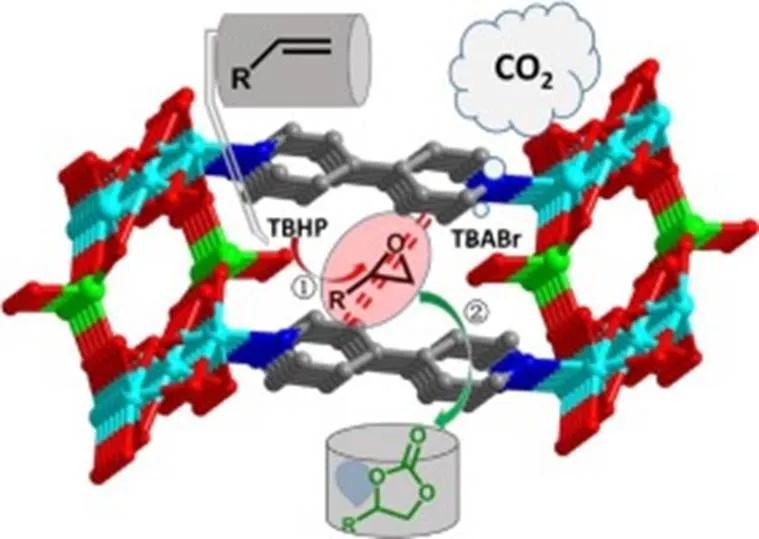
图10 CuMo-BPY催化烯烃合成环碳酸酯[53]
然而,不同于MOFs多功能催化,Nguyen等[55]只利用MOFs上的金属活性位点,同时实现了催化环氧化与环加成反应(见图11)。具体地,他们合成一系列的镧系金属有机框架材料(Ln-MOFs)MOF-590,MOF-591,MOF-592用于催化该反应。实验表明,MOF-590的催化效果最佳,其对TBHP和环氧化物均具有较强的化学吸附作用,MOF-590上活性Nd团簇可与TBHP作用,促进烯烃的环氧化;另外,Nd团簇Lewis中心与环氧化物的氧原子相互作用,协同Bu4NBr促进环氧化物开环以及生成环碳酸酯(产率高达91%)。
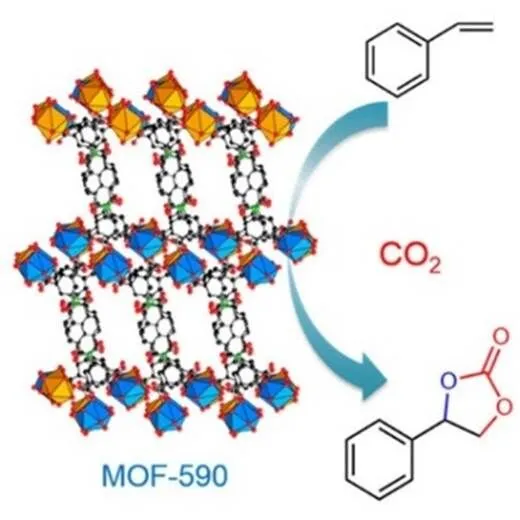
图11 MOF-590催化烯烃反应直接合成环碳酸酯[55]
6 其他催化剂
以烯烃为原料,催化其与CO2一步合成环碳酸酯的不同类型的催化剂被陆续开发出来,除以上催化剂外,贵金属及其化合物[56-59]、分子筛等也被用于催化烯烃环氧化-环加成反应合成环碳酸酯反应。如图12所示,负载型纳米Au/R201树脂[56]催化反应过程中,烯烃环氧化反应在纳米金表面发生,在季铵盐作用下,生成的环氧化物中间体与被活化的CO2发生环加成反应生成环碳酸酯,在该催化体系中,纳米金与季铵盐的协同作用是烯烃成功转化成环碳酸酯的关键。
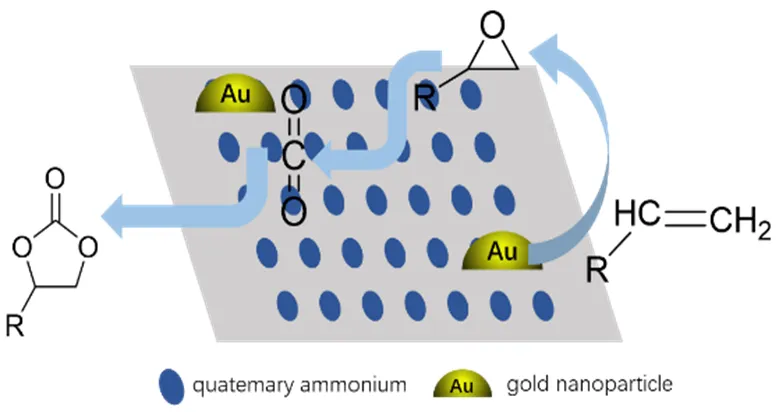
图12 Au/R201催化烯烃环氧化羧化生成环碳酸酯[56]
Srivastava等[60]以硅钛分子筛(TS-1或者TiMCM-41)为催化剂,DMAP为助催化剂分两步催化烯烃转化为环碳酸酯。当TS-1为催化剂,H2O2为氧化剂时,苯乙烯环碳酸酯的收率为6%左右;以TiMCM-41为催化剂,TBHP为氧化剂,苯乙烯环碳酸酯的产率可达33%。随后,Maksimchuk等[61]同样以TBHP为氧化剂,使用硅钛分子筛Ti-MMM-E/TBAB催化该反应,苯乙烯环碳酸酯的产率明显提高,可达66%。Huang等[62]则以绿色环保的分子氧为氧化剂,将不同过渡金属(Zn2+、Mn2+、Cr2+、Co2+、Fe3+等)掺杂到g-C3N4/SBA-15中合成复合型分子筛催化剂,用于催化苯乙烯到环碳酸酯的直接转化。结果表明,Fe-g-C3N4/SBA-15的催化活性最优,苯乙烯环碳酸酯的产率约32%(见图13)。
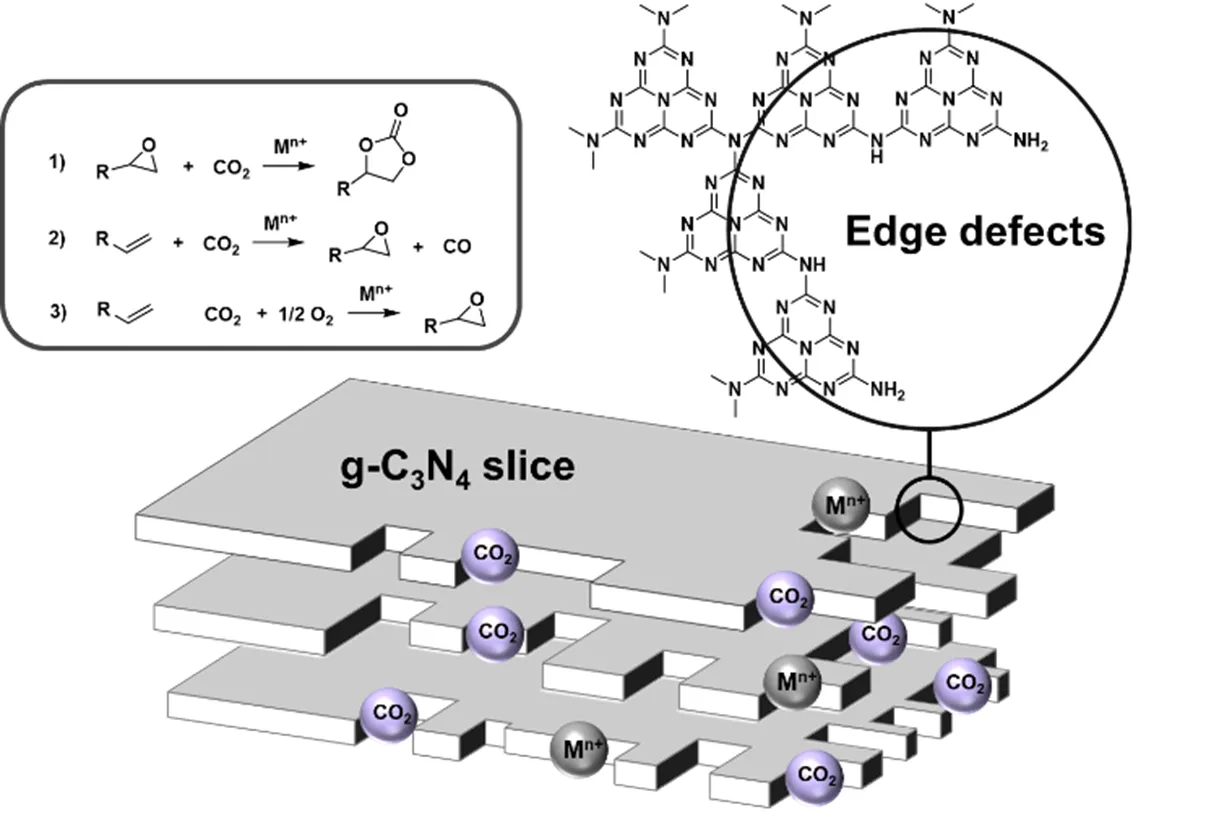
图13 Fe-g-C3N4/SBA-15催化烯烃-O2-CO2反应制备环碳酸酯[62]
Gu等[63]设计合成了类核桃分子筛LZ-276,将该类核桃分子筛协同KI催化苯乙烯环氧化-环加成反应,以TBHP为氧化剂,CO2压力为0.5 MPa,温度140 ℃,反应10 h,苯乙烯环碳酸酯产率为77%。若反应体系中不加KI时,无目标产物生成,说明KI在该体系中具有重要的作用。基于实验结果,作者还提出LZ-276/TBHP-KI和LZ-276/KI分别催化环氧化和环加成反应的发生,如图14所示,I−经TBHP氧化生成的IO−使苯乙烯碘化产生碘醇,碘醇进一步脱氢得到环氧化物中间体,随之类核桃分子筛LZ-276提供的铝金属位点与阴离子I−协同催化环氧化物中间体与CO2进行环加成反应,最终得到苯乙烯环碳酸酯。
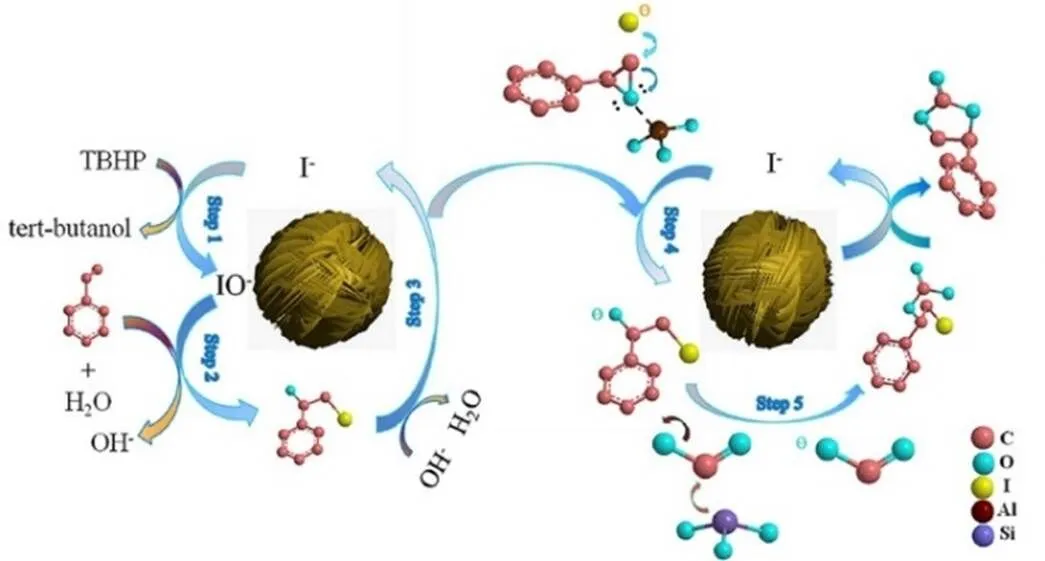
图14 LZ-276/KI催化苯乙烯与CO2合成环碳酸酯可能的反应机理[63]
电化学合成法是一种热门的合成策略。2013年,Gao等[64]对水相中电化学烯烃转化为环碳酸酯的方法进行了研究。如图15所示,该电化学反应体系不涉及贵金属和阳极的消耗,以Bu4NI/DMSO作为电解液,C-Ni作为电极,可以实现苯乙烯环碳酸酯高达94%的产率。反应发生的关键在于NH4+/NH3和I2/I−的氧化-还原循环过程,在电化学原位生成的I2和NH3的协同作用下,烯烃与CO2发生反应,生成环碳酸酯。
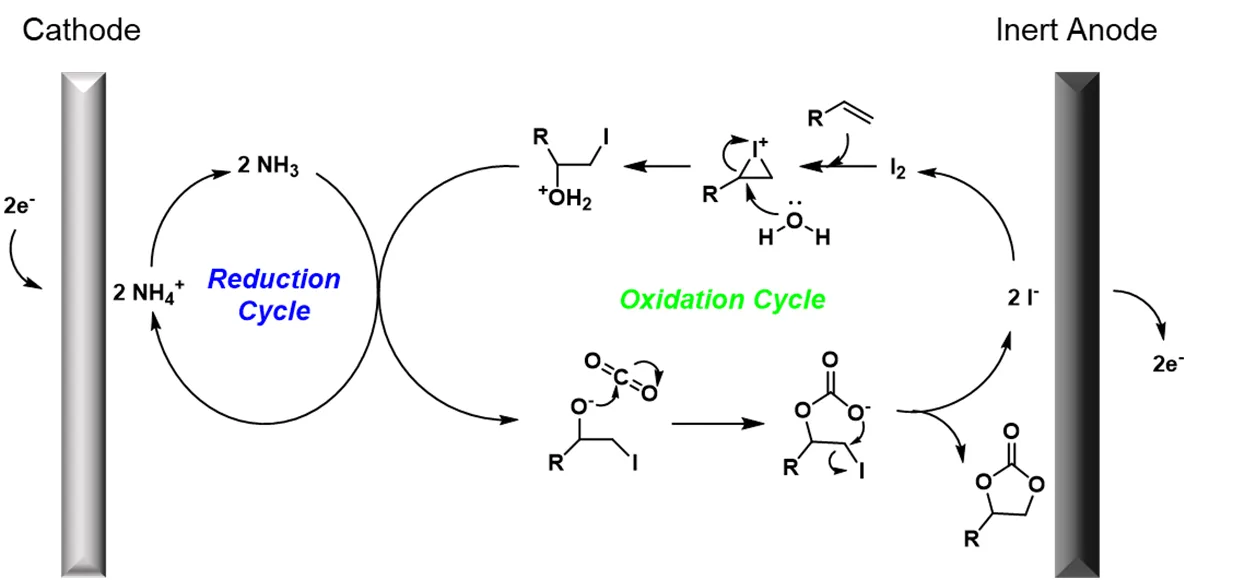
图15 电化学法促进烯烃与CO2直接合成环碳酸酯[64]
7 结 论
综上所述,从环氧化物的上游原料烯烃出发,与CO2经环氧化-环加成反应一步制备环碳酸酯不仅能降低原料成本,还能精简工艺,可极大提升反应经济性与环境友好性,对传统工艺革新具有重大的意义。但该反应路线目前还处于实验室初级阶段,新型高效催化体系还有待于开发。基于一步法合成环碳酸酯的实质,开发同时具备环氧化反应催化位点和环加成反应催化位点的催化体系是实现烯烃转化成环碳酸酯的关键。从多功能催化和产品分离的角度考虑,多功能化的非均相催化体系应用到该反应体系更适合未来化工工业发展。分子筛既是优良的催化剂,也是常见的载体,若能将多种催化位点构建到分子筛中,并在机理上避免环氧化反应与环加成反应相互抑制,达到烯烃到环碳酸酯高效转化的同时,催化剂具有较高的稳定性且良好的循环使用性能,对环碳酸酯合成行业的未来发展具有重大意义。
[1] SOLANGI K H, ISLAM M R, SAIDUR RA review on global solar energy policy [J]. Renewable & Sustainable Energy Reviews, 2011, 15(4): 2149-2163.
[2] DABRAL S, SCHAUB T. The use of carbon dioxide (CO2) as a building block in organic synthesis from an industrial perspective [J]. Advanced Synthesis & Catalysis, 2019, 361(2): 223-246.
[3] ALVES M, GRIGNARD B, MEREAU ROrganocatalyzed coupling of carbon dioxide with epoxides for the synthesis of cyclic carbonates: catalyst design and mechanistic studies [J]. Catalysis Science & Technology, 2017, 7(13): 2651-2684.
[4] 桂新胜, 曹发海, 刘殿华等超临界条件下二氧化碳与甲醇直接合成碳酸二甲酯 [J]. 高校化学工程学报, 1998, 12(2): 152-156.
GUI X S, CAO F H, LIU D H,. Synthesis of dimethyl carbonate from carbon dioxide under supercritical condition [J]. Journal of Chemical Engineering of Chinese Universities, 1998, 12(2): 152-156.
[5] HAN Q X, QI B, REN W MPolyoxometalate-based homochiral metal-organic frameworks for tandem asymmetric transformation of cyclic carbonates from olefins [J]. Nature Communications, 2015, 6: 1-8.
[6] CHEN F W, DONG T, XU T GDirect synthesis of cyclic carbonates from olefins and CO2catalyzed by a MoO2(acac)2-quaternary ammonium salt system [J]. Green Chemistry, 2011, 13(9): 2518-2524.
[7] EGHBALI N, LI C J. Conversion of carbon dioxide and olefins into cyclic carbonates in water [J]. Green Chemistry, 2007, 9(3): 213-215.
[8] TEBANDEKE E, COMAN C, GUILLOIS KEpoxidation of olefins with molecular oxygen as the oxidant using gold catalysts supported on polyoxometalates [J]. Green Chemistry, 2014, 16(3): 1586-1593.
[9] SUTTIKUL T, PAOSOMBAT B, SANTIKUNAPORN MImprovement of ethylene epoxidation in a parallel plate dielectric barrier discharge system by ethylene/oxygen separate feed and Ag catalyst [J]. Industrial & Engineering Chemistry Research, 2014, 53(10): 3778-3786.
[10] SUN J S, LIANG L, SUN J MDirect synthetic processes for cyclic carbonates from olefins and CO2[J]. Catalysis Surveys from Asia, 2011, 15(1): 49-54.
[11] WELTON T. Room-temperature ionic liquids. Solvents for synthesis and catalysis [J]. Chemical Reviews, 1999, 99(8): 2071-2083.
[12] HU J, MA J, LIU HDual-ionic liquid system: an efficient catalyst for chemical fixation of CO2to cyclic carbonates under mild conditions [J]. Green Chemistry, 2018, 20(13): 2990-2994.
[13] HAN L, CHOI S J, PARK M SCarboxylic acid functionalized imidazolium-based ionic liquids: Efficient catalysts for cycloaddition of CO2and epoxides [J]. Reaction Kinetics Mechanisms and Catalysis, 2012, 106(1): 25-35.
[14] DAI W L, JIN B, LUO S LCross-linked polymer grafted with functionalized ionic liquid as reusable and efficient catalyst for the cycloaddition of carbon dioxide to epoxides [J]. Journal of CO2Utilization, 2013, 3/4: 7-13.
[15] DAI W L, CHEN L, YIN S FHigh-efficiency synthesis of cyclic carbonates from epoxides and CO2over hydroxyl ionic liquid catalyst grafted onto cross-linked polymer [J]. Catalysis Letters, 2010, 137(1/2): 74-80.
[16] SUN J M, FUJITA S, BHANAGE B MOne-pot synthesis of styrene carbonate from styrene in tetrabutylammonium bromide [J]. Catalysis Today, 2004, 93/94/95: 383-388.
[17] SUN J M, FUJITA S, BHANAGE B MDirect oxidative carboxylation of styrene to styrene carbonate in the presence of ionic liquids [J]. Catalysis Communications, 2004, 5(2): 83-87.
[18] ZALOMAEVA O V, MAKSIMCHUK N V, CHIBIRYAEV A MSynthesis of cyclic carbonates from epoxides or olefins and CO2catalyzed by metal-organic frameworks and quaternary ammonium salts [J]. Journal of Energy Chemistry, 2013, 22(1): 130-135.
[19] LIU J, YANG G Q, LIU YMetal-free imidazolium hydrogen carbonate ionic liquids as bifunctional catalysts for the one-pot synthesis of cyclic carbonates from olefins and CO2[J]. Green Chemistry, 2019, 21(14): 3834-3838.
[20] GIRARD A L, SIMON N, ZANATTA MInsights on recyclable catalytic system composed of task-specific ionic liquids for the chemical fixation of carbon dioxide [J]. Green Chemistry, 2014, 16(5): 2815-2825.
[21] ARESTA M, DIBENEDETTO A, TOMMASI I. Direct synthesis of organic carbonates by oxidative carboxylation of olefins catalyzed by metal oxides: Developing green chemistry based on carbon dioxide [J]. Applied Organometallic Chemistry, 2000, 14(12): 799-802.
[22] WANG J L, WANG J Q, HE L NA CO2/H2O2-tunable reaction: Direct conversion of styrene into styrene carbonate catalyzed by sodium phosphotungstate/n-Bu4NBr [J]. Green Chemistry, 2008, 10(11): 1218-1223.
[23] XIE J N, DIAO Z F, QIAO COne-pot stepwise synthesis of cyclic carbonates directly from olefins with CO2promoted by K2S2O8/NaBr [J]. Journal of CO2Utilization, 2016, 16: 313-317.
[24] ZHANG S, XIA Z M, ZOU YInterfacial frustrated lewis pairs of CeO2activate CO2for selective tandem transformation of olefins and CO2into cyclic carbonates [J]. Journal of the American Chemical Society, 2019, 141(29): 11353-11357.
[25] WANG H L, GUO L P. Mechanistic Insights into cycloaddition of CO2with epoxide catalyzed by a bimetallic (salen)Fe(II)Cl2complex with/without a cocatalyst [J]. ChemistrySelect, 2020, 5(8): 2516-2521.
[26] DARENSBOURG D J. Synthetic biodegradable polymers [M]. Berlin: Springer, 2012.
[27] CHEN Y J, LUO R C, XU Q HCharged metalloporphyrin polymers for cooperative synthesis of cyclic carbonates from CO2under ambient conditions [J]. ChemSusChem, 2017, 10(11): 2534-2541.
[28] BAI D S, ZHANG Z Y, WANG G JCycloaddition of epoxide and CO2to cyclic carbonate catalyzed by VO(IV) porphyrin [J]. Applied Organometallic Chemistry, 2015, 29(4): 240-243.
[29] KUMAR S, JAIN S L, SAIN B. Metal acetylacetonates as highly efficient and cost effective catalysts for the synthesis of cyclic carbonates from CO2and epoxides [J]. Catalysis Letters, 2012, 142(5): 615-618.
[30] GROVES J T, QUINN R. Aerobic epoxidation of olefins with ruthenium porphyrin catalysts [J]. Journal of the American Chemical Society, 1985, 107(20): 5790-5792.
[31] LANE B S, BURGESS K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide [J]. Chemical Reviews, 2003, 103(7): 2457-2473.
[32] MCGARRIGLE E M, GILHEANY D G. Chromium- and manganese-salen promoted epoxidation of alkenes [J]. Chemical Reviews, 2005, 105(5): 1563-1602.
[33] ZHANG W, LOEBACH J L, WILSON S REnantioselective epoxidation of unfunctionalized olefins catalyzed by (salen)manganese complexes [J]. Journal of the American Chemical Society, 1990, 112(7): 2801-2803.
[34] KUMAR S, SINGHAL N, SINGH R KDual catalysis with magnetic chitosan: Direct synthesis of cyclic carbonates from olefins with carbon dioxide using isobutyraldehyde as the sacrificial reductant [J]. Dalton Transactions, 2015, 44(26): 11860-11866.
[35] DIAS L D, CARRILHO R M B, HENRIQUES C AA recyclable hybrid manganese(III) porphyrin magnetic catalyst for selective olefin epoxidation using molecular oxygen [J]. Journal of Porphyrins and Phthalocyanines, 2018, 22(4): 331-341.
[36] HE L, NATH J K, LIN Q P. Robust multivariate metal-porphyrin frameworks for efficient ambient fixation of CO2to cyclic carbonates [J]. Chemical Communications, 2019, 55(3): 412-415.
[37] HASEGAWA J, MIYAZAKI R, MAEDA CTheoretical study on highly active bifunctional metalloporphyrin catalysts for the coupling reaction of epoxides with carbon dioxide [J]. Chemical Record, 2016, 16(5): 2260-2267.
[38] BAI D S, JING H W. Aerobic oxidative carboxylation of olefins with metalloporphyrin catalysts [J]. Green Chemistry, 2010, 12(1): 39-41.
[39] DIAS L D, CARRILHO R M B, HENRIQUES C AHybrid metalloporphyrin magnetic nanoparticles as catalysts for sequential transformation of alkenes and CO2into cyclic carbonates [J]. ChemCatChem, 2018, 10(13): 2792-2803.
[40] MONDAL I, CHATTOPADHYAY S. Development of multi-metallic complexes using metal-salen complexes as building blocks [J]. Journal of Coordination Chemistry, 2019, 72(19/20/21): 3183-3209.
[41] GUALANDI A, CALOGERO F, POTENTI SAl(salen) metal complexes in stereoselective catalysis [J]. Molecules, 2019, 24(9), 1716.
[42] DALTON C T, RYAN K M, WALL V MRecent progress towards the understanding of metal-salen catalysed asymmetric alkene epoxidation [J]. Topics in Catalysis, 1998, 5(1/2/3/4): 75-91.
[43] QIU J P, YU L, NI J GPalladium-salen-bridged ionic networks immobilized on magnetic dendritic silica fibers for the synthesis of cyclic carbonates by oxidative carboxylation [J]. New Journal of Chemistry, 2020, 44(4): 1269-1277.
[44] KIM H, MOON H-S, SOHAIL MSynthesis of cyclic carbonate by CO2fixation to epoxides using interpenetrated MOF-5/n-Bu4NBr [J]. Journal of Materials Science, 2019, 54(18): 11796-11803.
[45] KIM J, KIM S-N, JANG H-GCO2cycloaddition of styrene oxide over MOF catalysts [J]. Applied Catalysis A-General, 2013, 453: 175-180.
[46] SONG J, ZHANG Z, HU SMOF-5/n-Bu4NBr: An efficient catalyst system for the synthesis of cyclic carbonates from epoxides and CO2under mild conditions [J]. Green Chemistry, 2009, 11(7): 1031-1036.
[47] XU K, MOELJADI A M P, BINH KHANH MHow does CO2react with styrene oxide in Co-MOF-74 and Mg-MOF-74? Catalytic mechanisms proposed by QM/MM calculations [J]. Journal of Physical Chemistry C, 2018, 122(1): 503-514.
[48] XUE Z, JIANG J, MA M-GGadolinium-based metal-organic framework as an efficient and heterogeneous catalyst to activate epoxides for cycloaddition of CO2and alcoholysis [J]. ACS Sustainable Chemistry & Engineering, 2017, 5(3): 2623-2631.
[49] BROWN K, ZOLEZZI S, AGUIRRE PCu(H2btec)(bipy)∞: A novel metal organic framework (MOF) as heterogeneous catalyst for the oxidation of olefins [J]. Dalton Transactions, 2009(8): 1422-1427.
[50] CANCINO P, PAREDES-GARCIA V, AGUIRRE PA reusable Cu-II based metal-organic framework as a catalyst for the oxidation of olefins [J]. Catalysis Science & Technology, 2014, 4(8): 2599-2607.
[51] CHEN J W, CHEN M D, ZHANG B YAllylic oxidation of olefins with a manganese-based metal-organic framework [J]. Green Chemistry, 2019, 21(13): 3629-3636.
[52] TANG J, DONG W J, WANG GEfficient molybdenum(VI) modified Zr-MOF catalysts for epoxidation of olefins [J]. RSC Advances, 2014, 4(81): 42977-42982.
[53] SHI Z L, NIU G Q, HAN Q XA molybdate-incorporated cooperative catalyst: High efficiency in the assisted tandem catalytic synthesis of cyclic carbonates from CO2and olefins [J]. Molecular Catalysis, 2018, 461: 10-18.
[54] KE S-C, LUO T-T, CHANG G-GSpatially ordered arrangement of multifunctional sites at molecule level in a single catalyst for tandem synthesis of cyclic carbonates [J]. Inorganic Chemistry, 2020, 59(3): 1736-1745.
[55] NGUYEN H T D, TRAN Y B N, NGUYEN H NA series of metal-organic frameworks for selective CO2capture and catalytic oxidative carboxylation of olefins [J]. Inorganic Chemistry, 2018, 57(21): 13772-13782.
[56] XIANG D, LIU X F, SUN J SA novel route for synthesis of styrene carbonate using styrene and CO2as substrates over basic resin R201 supported Au catalyst [J]. Catalysis Today, 2009, 148(3/4): 383-388.
[57] WANG Y L, SUN J H, XIANG DA facile, direct synthesis of styrene carbonate from styrene and CO2catalyzed by Au/Fe(OH)3-ZnBr2/Bu4NBr system [J]. Catalysis Letters, 2009, 129(3/4): 437-443.
[58] SUN J M, FUJITA S I, ZHAO F YA direct synthesis of styrene carbonate from styrene with the Au/SiO2-ZnBr2/Bu4NBr catalyst system [J]. Journal of Catalysis, 2005, 230(2): 398-405.
[59] JASIAK K, KRAWCZYK T, PAWLYTA MOne-pot synthesis of styrene carbonate from styrene and CO2over the nanogold-ionic liquid catalyst [J]. Catalysis Letters, 2016, 146(5): 893-901.
[60] SRIVASTAVA R, SRINIVAS D, RATNASAMY P. Synthesis of polycarbonate precursors over titanosilicate molecular sieves [J]. Catalysis Letters, 2003, 91(1/2): 133-139.
[61] MAKSIMCHUK N V, IVANCHIKOVA I D, AYUPOV A BOne-step solvent-free synthesis of cyclic carbonates by oxidative carboxylation of styrenes over a recyclable Ti-containing catalyst [J]. Applied Catalysis B-Environmental, 2016, 181: 363-370.
[62] HUANG Z J, LI F B, CHEN B FWell-dispersed g-C3N4nanophases in mesoporous silica channels and their catalytic activity for carbon dioxide activation and conversion [J]. Applied Catalysis B-Environmental, 2013, 136: 269-277.
[63] GU B B, XU T, XU G RSynthesis of styrene carbonate from styrene and CO2catalyzed by walnut-like zeolite LZ-276 [J]. Microporous and Mesoporous Materials, 2020, 293: 109779.
[64] GAO X F, YUAN G Q, CHEN H JEfficient conversion of CO2with olefins into cyclic carbonates via a synergistic action of I2and base electrochemically generated in situ [J]. Electrochemistry Communications, 2013, 34: 242-245.
Progress on direct synthesis of cyclic carbonates from olefins and CO2
YI Min1,2, CHEN Jing-wen1,2, TONG Yang3, BAO Zong-bi1,2, YANG Qi-wei1,2,YANG Yi-wen1,2, ZHANG Zhi-guo1,2
(1. Key Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China;2. Institute of Zhejiang University-Quzhou, Quzhou 324000, China 3. High-Tech Research and Development Center, Ministry of Science and Technology of the People's Republic of China, Beijing 100044, China)
CO2is a cheap and abundant C1 resource. Chemical fixation of CO2into cyclic carbonates is one of the most promising routes for CO2utilization in industry. Olefins are low-toxic and inexpensive when compared with epoxides. Cyclic carbonates can be prepared in one step by a tandem olefin epoxidation and cycloaddition of CO2using olefins as starting materials. This reaction is a potential synthetic route in industry in terms of short reaction steps and high atomic economy. This review summarizes recent advances in direct synthesis of cyclic carbonates from olefins and CO2emphasized on different catalysts reported, i.e. ionic liquids (ILs), metal oxides or metal salts, metal organic complexes and metal-organic frameworks (MOFs). Perspectives in this field are discussed.
carbon dioxide; olefins; one-step synthesis; cyclic carbonates
TQ032
A
10.3969/j.issn.1003-9015.2021.01.003
1003-9015(2021)01-0024-10
2020-04-09;
2020-05-28。
国家自然科学基金(21878266,22078288);浙江大学衢州研究院科研基金(IZQ2019-KJ-001)。
易敏(1994-),女,江西萍乡人,浙江大学硕士生。
杨亦文,E-mail:ceywyang@zju.edu.cn
猜你喜欢
科学大众(2023年17期)2023-10-26 07:38:56
小天使·二年级语数英综合(2021年5期)2021-07-11 10:58:35
中学化学(2019年4期)2019-08-06 13:59:37
中学化学(2019年4期)2019-08-06 13:59:37
浙江农业学报(2017年1期)2017-05-17 06:13:45
中学化学(2017年2期)2017-04-01 08:51:54
化工进展(2015年6期)2015-11-13 00:29:40
世界热带农业信息(2014年11期)2015-01-05 17:19:22
中成药(2014年11期)2014-02-28 22:29:49
无机化学学报(2014年7期)2014-02-28 17:32:10
