
光激发金属配位四苯基卟啉瞬态吸收和衰减动力学性质研究
2021-03-18 15:10马子辉王梦妍曹洪玉王立皓郑学仿
高等学校化学学报 2021年3期
马子辉,王梦妍,曹洪玉,唐 乾,王立皓,郑学仿
(1.大连大学生命科学与技术学院,2.环境与化学工程学院,大连116622)
叶绿素和血红素(铁卟啉)等金属卟啉化合物对生命体内光合作用、输氧、储氧及电子传递等过程起着至关重要的作用[1].光合作用中叶绿素是光能转换的反应中心,血红素类蛋白能够在光诱导下发生氧化还原反应[2].Sakai等[3]发现365 nm氙灯光能诱导高铁血红蛋白(metHb)还原生成碳氧血红蛋白(HbCO),推测电子可能是从卟啉环转移至卟啉环中心铁;Gu等[4]发现醇的加入有利于血红素类蛋白的光还原.紫外光或可见光激发色氨酸对其它生物分子会有电子转移反应影响[5],实验表明游离色氨酸受光激发后可以发生能量转移到铁卟啉中,引起蛋白光谱变化[6].以上研究表明光对金属卟啉均有明显的反应重要引发作用,其中确定光反应中间体是理解反应的关键.光激发金属卟啉至激发态是光合作用反应的第一步.过渡金属配位卟啉分子具有大共轭结构,在吸收光后其激发态具有复杂性和多样性,从而具备优异的光化学和光物理性质[7],因此在金属有机骨架(MOFs)材料[8]、染料太阳能电池、光动力学治疗光敏剂、光催化及光能转化材料科学等领域引起广泛关注.Steven等[9]在其实验条件下表征了四苯基卟啉的时间分辨共振拉曼光谱,通过同位素位移技术与各谱带的偏振系数,准确地归属了四苯基卟啉的基态和激发态在共振拉曼光谱上的信号.Seiji等[10]根据卟啉的激发态弛豫过程推测出卟吩激发态的衰变过程经历B带内转换到Qy带和Qx带;Qx激发态与溶剂发生能量交换振动弛豫后再经过衰变12 ns返回基态,并根据卟啉的激光激发后的短时激发性质对光谱进行归属.吴骊珠等[11]以四苯基卟啉铂为光敏剂构建二氧化硅纳米颗粒的三重态-三重态湮灭上转换体系,光敏剂所占比例小(1∶40),效率较高,光敏剂吸收低能量的光子跃迁至其单重激发态并通过系间窜越过程到达其三重激发态,随后处于三重激发态的光敏剂通过能量传递将其三重态能量传递给发光体分子.
血红素和叶绿素分别以铁和镁为金属活性中心,卟啉光敏剂的研究倾向于采用锌或镍等过渡态金属,选择原因均与金属配位性质相关,但尚缺乏光激发实验和理论数据;光激发金属卟啉氧化还原反应中长时中间态是反应基础,相关实验报道较少,致使其机理也尚缺乏解析.叶绿素和铁卟啉侧链过长,不利于金属卟啉核心部位的光学性质研究,本文以四苯基卟啉为模板分子,采用稳态吸收光谱和激光闪光光解方法,以间-四苯基卟吩(TPP-2H),间-四苯基卟啉氯化铁(Ⅲ)(TPP-FeCl),5,10,15,20-四苯基卟啉-21H,23H-卟吩镍(Ⅱ)(TPP-Ni),5,10,15,20-四苯基卟啉-21H,23H-卟吩镁(TPP-Mg)和5,10,15,20-四苯基卟啉-21H,23H-卟吩锌(TPP-Zn)5个化合物为模型研究光激发后卟啉衍生物的光学性质,归属谱峰,分析5种卟啉化合物在光激发前后的特点与差异机理,发现并理解金属配位对卟啉光学性质的影响,进而协助理解光激发的光合作用或血红素氧化还原反应.
1 实验部分
1.1 试剂与仪器
TPP-FeCl(分析纯)购自上海阿法埃莎(中国)化学有限公司;TPP-Ni、TPP-Mg、TPP-2H(纯度>99%,TPP-2H,无氯)和二甲基亚砜(纯度>99.9%,DMSO)均为分析纯,购自上海阿拉丁生化科技股份有限公司;TPP-Zn(纯度95%)购自北京百灵威科技有限公司.经紫外-可见光谱检测TPP-FeCl中均是三价铁.
Jasco-V-560型紫外-可见分光光度计和FP 6500型荧光分光光度计(日本分光株式会社);LP980型激光闪光光解仪(英国爱丁堡仪器公司).
1.2 实验方法
紫外-可见光谱和荧光光谱测定:将5×10-6mol/L TPP-FeCl,TPP-Mg,TPP-Ni,TPP-Zn和TPP-2H的DMSO溶液分别置于比色皿中,采用紫外-可见分光光度计测量样品激光照射前后300~700 nm的紫外-可见吸收光谱,狭缝宽度2 nm,扫描速度400 nm/min;使用荧光分光光度计测定激发波长为355 nm的发射光谱,测定范围380~420 nm,激发光和发射光狭缝宽度均为3 nm.
激发态瞬态光谱和动力学测定:将5×10-6mol/L TPP-FeCl,TPP-Mg,TPP-Ni,TPP-Zn和TPP-2H的DMSO溶液分别置于石英比色皿中,经过固定波长(355 nm)的激光发射器激发,测量其瞬态吸收的变化及衰减曲线.采用激光闪光光解仪测量,激光模式1 Hz;检测时间范围为4000 ns;每次测量2次激光脉冲.检测器狭缝宽度为2 nm;检测波长范围360~750 nm.在获取激光瞬态吸收光谱之后,进行谱图分析并将捕捉到的谱图进行截取,截取0~2000 ns数据,谱线平均宽度为10 ns.其它参数设为默认数值.

式中:ΔOD(a.u.)为激发态和基态的光密度差值;εt和εG(L·mol-1·cm-1)分别为瞬态中间体和基态的消光系数;c(mol/L)为基态分子转移到瞬态中间体的浓度,d(cm)为探测光束的有效光程长度[12].激光闪光光解检测的ΔOD(a.u.)值是时间(t)和波长(λ)两个变量的函数:

式中:εT(L·mol-1·cm-1)为激发态的消光系数;cT(mol/L)为三重激发态的浓度;cs(mol/L)为单重激发态的浓度;φ(a.u.)为样品的荧光光谱强度.测定过程中采用定时或定波长转为单变量函数.激发态衰减动力学拟合曲线采用下式:

式中:Bi和τi分别为指前因子和特征寿命;A为附加背景.R(t)通常被称为样品衰减模型,其是样品对无限短激发响应的理论表达式,实验以ΔOD值作为R(t)值进行动力学分析,拟合曲线采用卡方检验方法,在最低χ2值时确定最佳拟合曲线各参数.
1.3 理论计算
TPP-2H和金属配位卟啉结构的理论计算采用密度泛函理论在Gaussian 09程序包[13]中完成,几何构型均采用已经证实卟啉类分子计算与实验结果吻合的B3LYP/lanl2dz基组水平优化[14,15],采用开壳层UB3LYP/lanl2dz基组对TPP-FeCl进行优化,频率分析无虚频,表明优化的结果是稳定的构型;采用含时密度泛函理论在B3LYP/lanl2dz基组下进行激发态计算,TPP-FeCl采用开壳层UB3LYP/lanl2dz基组水平计算,激发态数量设置为240.基态和激发态计算结果利用Gausssum3.0软件[16]对前线轨道和电子吸收光谱进行分析.
2 结果与讨论
2.1 金属卟啉激发前后稳态吸收光谱
5种卟啉化合物TPP-2H,TPP-FeCl,TPP-Ni,TPP-Mg和TPP-Zn的差异仅在于中心原子不同(图1),但卟啉化合物的稳态紫外吸收谱图差异较大[图2(A)~(E)].光激发前,TPP-2H紫外吸收谱中Soret带有一个较强吸收峰,Q带有4个弱吸收峰,金属卟啉TPP-FeCl,TPP-Ni,TPP-Mg和TPP-Zn的Soret带最大吸收峰分别位于414,416,426和428 nm处[图2(B)~(E)].
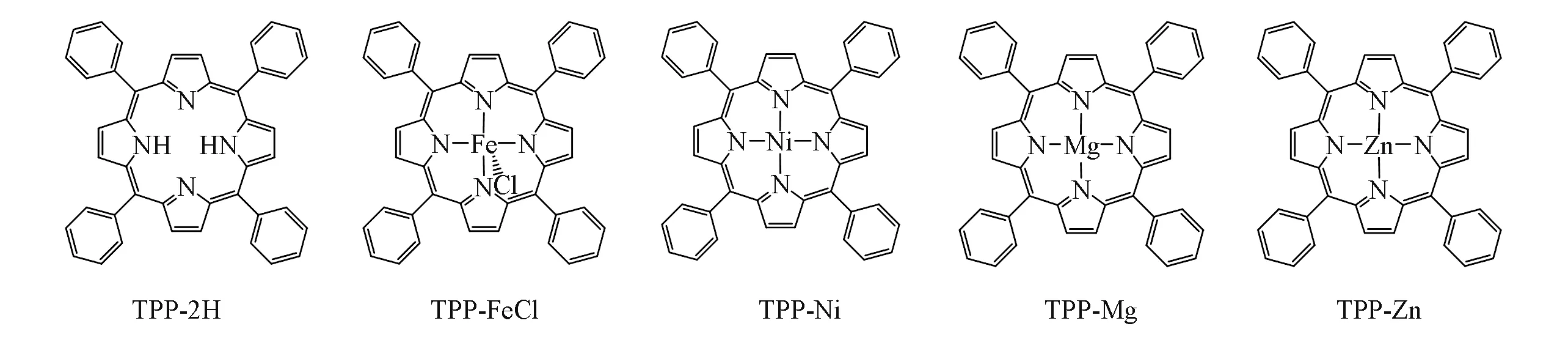
Fig.1 Structures of tetraphenylporphyrin and metal-coordinated tetraphenylporphyrins
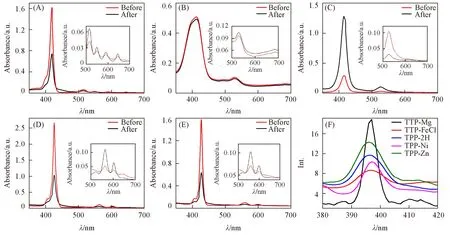
Fig.2 UV-Vis spectra of TPP-2H(A),TPP-FeCl(B),TPP-Ni(C),TPP-Mg(D),TPP-Zn(E)before and after 355 nm laser excitation and fluorescence spectra with 355 nm light excitation(F)
光激发次数较少时,各卟啉化合物稳态光谱性质稳定,紫外光谱基本不变.在受多次355 nm激光激发后,不同卟啉的光谱变化趋势和程度各有差异.经过激光照射,不含金属配体的TPP-2H在Soret带418 nm处最大吸收峰在光激发后明显降低,其降低程度与光激发TPP-Zn后在428 nm处最大吸收峰降低程度相同,说明这2种卟啉化合物受光激发后其共轭结构发生变化.TPP-Ni经光激发后,Soret带和Q带吸收强度均出现大幅上升,Soret带吸光度由0.3 a.u.升至1.3 a.u.;Q带吸光度由0.04 a.u.上升到0.11 a.u.,且峰位置没有发生变化.光激发后TPP-2H,TPP-Zn和TPP-Mg的Q带峰位置未发生变化但强度均明显降低,表明多次激光照射后原分子减少.TPP-FeCl的紫外吸收光谱Soret带和Q带谱峰强度降低均不明显,表明Fe与卟啉环的配位较为稳定,且不易受到激光激发影响.以上化合物光激发后长时间(>5 min)放置,其稳态光谱均不能恢复至激光照射前,说明光激发可使卟啉化合物的结构发生改变.
2.2 金属卟啉光激发瞬态吸收光谱
TPP-2H,TPP-FeCl,TPP-Ni,TPP-Mg和TPP-Zn的瞬态动力学吸收有显著差异(图3).5种卟啉化合物在396 nm处均有瞬态吸收光谱负峰.根据5种卟啉化合物在激发波长为355 nm时的发射光谱,396 nm处的谱峰为卟啉的荧光峰[图2(F)],根据稳态荧光和式(2),此负峰归属为卟啉荧光项[φ(λ)cs(t)].不同分子396 nm处的谱峰强度差异很大,其中TPP-FeCl最强,其ΔOD值达到-0.038 a.u.;TPP-2H的396 nm谱峰ΔOD值为-0.018 a.u.;TPP-Ni,TPP-Mg和TPP-Zn的谱峰强度均小于-0.001 a.u.,以上差异是受环内离子对TPP三重激发态稳定性影响.
TPP-FeCl*的瞬态吸收光谱中Soret带的ΔOD为0,即无明显谱峰,说明激光激发过程中,TPP-FeCl未产生中间态.TPP-2H,TPP-Ni,TPP-Mg和TPP-Zn的稳态紫外-可见吸收峰中Soret带谱峰位置在瞬态吸收光谱中也均出现明显负峰,根据式(1)可知,激光激发后中间体的εt明显小于基态的εG,导致ΔOD值为负值.瞬态吸收光谱中,Soret带负吸收峰在不同卟啉中明显不同,TPP-2H*和TPP-Zn*分别在418和428 nm的负峰较为显著,TPP-Ni*和TPP-Mg*分别在416和426 nm的Soret带负峰较弱(图3).
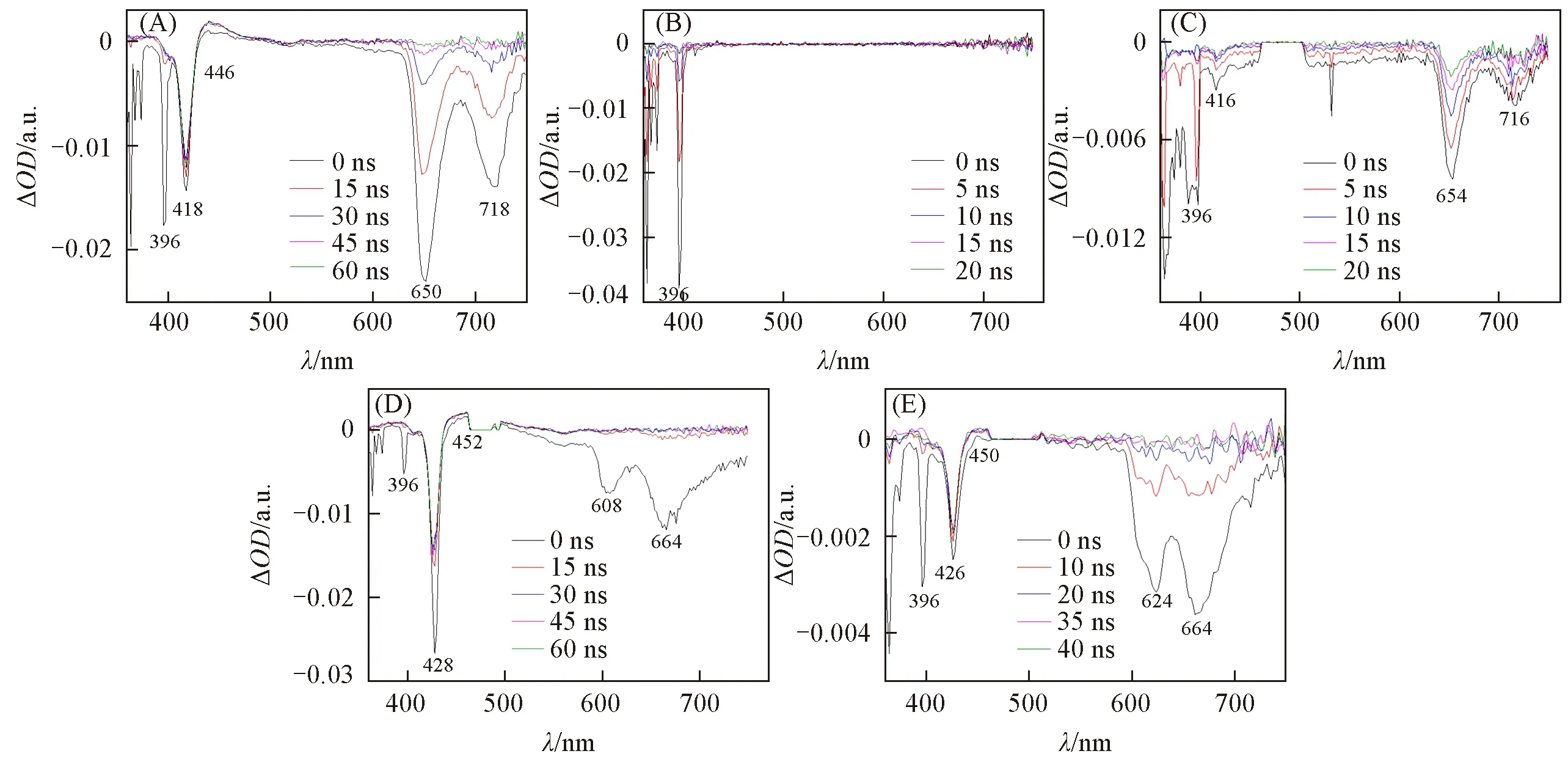
Fig.3 Transient absorption spectral maps of TPP-2H(A),TPP-FeCl(B),TPP-Ni(C),TPP-Zn(D)and TPPMg(E)photoexcited by 355 nm laser pulse
Soret带是分子被激发至第二激发单重态S2(0-0)跃迁的吸收峰,卟啉环中心原子或金属影响分子的电子排布和共轭结构[17],光谱表明氢和锌对光激发后分子内电子跃迁至S2变化影响较明显,ΔOD值变化较大.在TPP-2H*,TPP-Mg*和TPP-Zn*的瞬态吸收光谱中出现了3个正的弱峰,分别位于448,452和450 nm,与其各自的Soret带强负吸收峰位置接近,其中正吸收峰归属为激发态吸收εt大于基态吸收εG,说明3种卟啉均产生了三重激发态中间体.
吸收谱带可表明激发态特征[18].Q带吸收是最低能级单重激发态S1的电子态,TPP-2H,TPP-Mg和TPP-Zn在稳态吸收谱相应具有Q带吸收峰位置处均未发现瞬态吸收谱峰,表明基态和激发态的Q带吸收强度无差别,ΔOD值无变化,即S0→S1的跃迁未改变;TPP-Ni在524 nm处有明显负峰,表明激光激发后中间态改变了最低能级跃迁,中间体的εt明显小于基态的εG,导致ΔOD值为负值.TPP-2H*,TPP-Ni*,TPP-Mg*和TPP-Zn*的瞬态光谱中分别在650 nm/718 nm,654 nm/716 nm,608 nm/664 nm和624 nm/664 nm处有负峰,谱峰信号均呈现强度大、宽度大的特性,根据式(2),这些负峰信号为基态漂白峰,归属为基态吸收εG(λ)值.TPP-FeCl*的瞬态吸收光谱中Soret带和Q带无明显谱峰,这一显著差别表明Fe电子排布的独特性致使卟啉瞬时激发态中间体异于其它金属配位卟啉.
2.3 金属卟啉激发态瞬态动力学
对TPP-2H,TPP-FeCl,TPP-Ni,TPP-Mg和TPP-Zn的瞬态吸收光谱中各自特征吸收波长的激发态衰减曲线根据式(3)进行拟合(表1).5种卟啉化合物光激发下在396 nm负峰的瞬态衰减时长均相同,在5 ns内此峰消失,这是5种卟啉化合物中共有的瞬态衰减过程,根据此峰的特点,可以推测卟啉大环的共轭结构受到光激发后有相同能量弛豫路径产生,产生共同的荧光峰.此谱峰可以归属为卟啉大分子配体的S0→S1跃迁,但各种卟啉化合物在396 nm处的谱峰强度不同,说明此跃迁在不同卟啉之间的几率差异较大,也显示出分子处于激发态后能量弛豫途径不同.
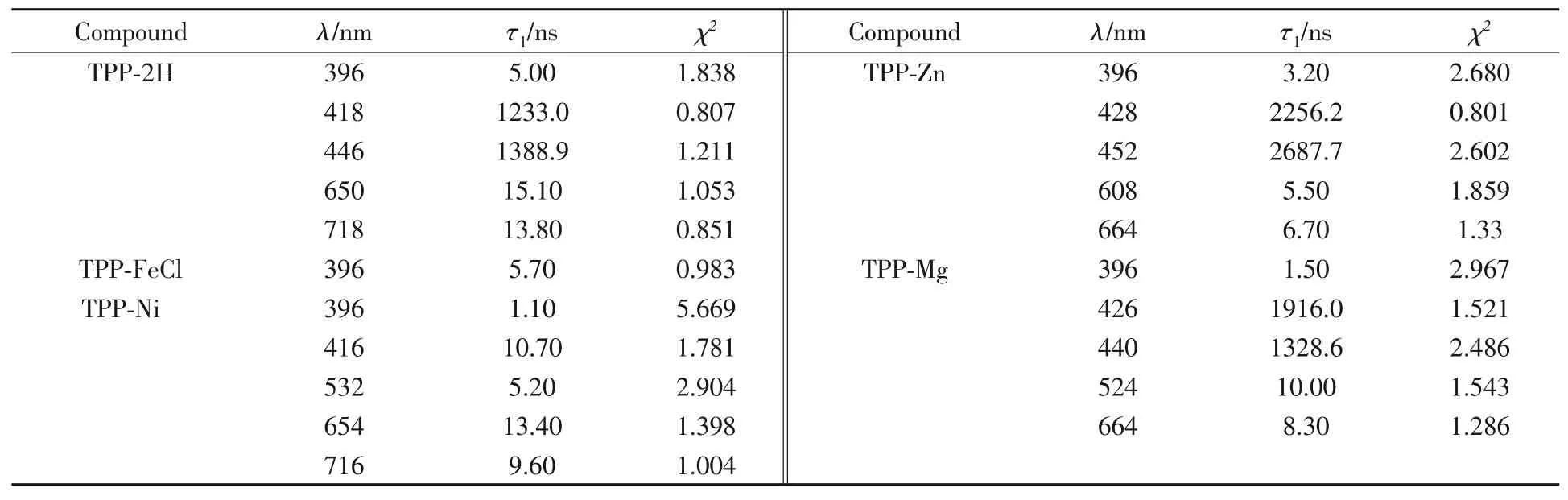
Table 1τ1,χ2 values of TPP-2H,TPP-FeCl,TPP-Ni,TPP-Mg and TPP-Zn
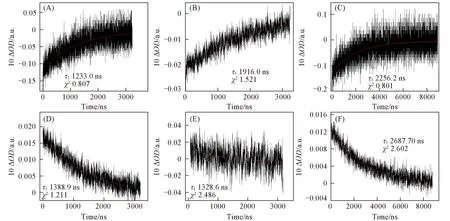
Fig.4 Long-life time Soret bands decay kinetic curves of excited porphyrin compounds after 355 nm laser-excitation
特征吸收波长的激发态衰减曲线拟合数据表明,金属配位卟啉三重激发态的正峰衰减过程中,TPP-2H*的446 nm处的峰衰减时长为1388.9 ns,TPP-Mg*在440 nm处的峰衰减时长为1328.6 ns,TPPZn*在452 nm处的峰瞬态衰减时长2687.7 ns,邻近Soret带418,426和428 nm负峰衰减动力学时长分别为1233.0,1916.0和2256.2 ns(图4).正峰和负峰的动力学时长均在同一数量级,具有极为相似的动力学性质,表明分子被光激发产生的新激发中间态、基态卟啉浓度减小,致使原Soret带ΔOD值呈现负峰;而在较长波长的正峰则表明此激发中间态有弱允许电子跃迁,有较低的摩尔吸光系数εt,产生正ΔOD峰.正峰和负峰同时出现及相同数量级的激发态动力学时长表明同一卟啉激发态共轭或配位结构状态改变时,其能级轨道和电子跃迁的改变情况.TPP-2H*,TPP-Mg*和TPP-Zn*较长时间的能量弛豫过程有利于能量累积,若在此时间间隔内进行二次激发,分子更有利于进入更高能级激发态,因而TPP-Zn可提供较好的光电转换效率.在可见光照射下,锌酞菁/氮化碳即可被激发用于光电催化CO2还原反应[19],原因可能为电子-空穴对的寿命长,它们参与反应的机会增大,这可能也是其在太阳能敏化剂等领域发挥着重要的作用原因之一[20,21].TPP-Ni在416 nm处的动力学时长非常短,仅为10.7 ns,且在较长波长位置并未出现正峰,显示了在Ni配位四苯基卟啉中,Ni提供的轨道和电子阻碍了类似于上述3种卟啉中间态结构的产生.分析分子结构及光谱规律,可以发现由于TPP-2H,TPP-Mg和TPP-Zn的内部氢和镁、锌离子最外层电子饱和,易于产生长时激发态中间体;铁离子提供空轨道和单电子,卟啉电子激发后易离域到铁离子中,难以形成激发态;镍离子最外层只提供一个空轨道,镍上电子与卟啉N配位,空轨道不与卟啉共平面,因而兼具二者性质.
TPP-2H*,TPP-Ni*,TPP-Mg*和TPP-Zn*瞬态光谱中的基态漂白峰650 nm/718 nm,654 nm/716 nm,624 nm/664 nm和608 nm/664 nm处的强负峰寿命较短,动力学寿命在5~15 ns之间,与396 nm波长处的负峰相似,归属为激光激发后产生的整个卟啉分子的强荧光谱峰[21~23],为Q带(0-0)跃迁产生.Q带(0-0)跃迁极易受卟啉内配位金属和氢原子影响,TPP-2H分子激发态通过系间窜越至三重态,在650和718 nm处产生较强荧光;Mg,Ni和Zn闭壳层金属配位的分子激发态通过系间窜越至T1态减弱,荧光峰强度降低;开壳层金属Fe配位激发态S1易于恢复至基态,Q带(0-0)跃迁的分子数过低,荧光峰难以检测到.
2.4 密度泛函理论和含时密度泛函理论计算
TPP-2H符合Gouterman四轨道理论[24],HOMO和LUMO之间的能级差为2.63 eV,4个前线轨道HOMO-1,HOMO,LUMO和LUMO+1轨道均位于与卟啉的4个吡咯环和N原子上,LUMO和LUMO+1轨道能级简并,HOMO-2及以下占据轨道能级均较低,远离前线轨道(图5).中心镁、锌和镍配位后卟啉分子HOMO和LUMO之间的能极差增大,LUMO和LUMO+1能级轨道简并.TPP-Mg和TPP-Zn的前线轨道基本分布在卟啉内部的4个吡咯环和meso位的C上,不脱离卟啉共轭结构,其稳态紫外-可见光吸收与TPP-2H有较大相似之处,Mg2+和Zn2+离子结构为满电子结构,金属离子与4个N原子相连,影响卟啉大共轭体系的n-π*跃迁,在卟啉分子内电子传递过程中起着重要作用,因而瞬态吸收峰二者差异较大.TPP-2H,TPP-Mg和TPP-Zn 3种卟啉分子的单重激发态的S0→S1和S0→S2跃迁,均为4个前线轨道,如TPP-Mg的S0→S1跃迁组成为HOMO-1→LUMO+1(36%)和HOMO→LUMO(64%),S0→S2跃迁组成为HOMO-1→LUMO(36%)和HOMO→LUMO+1(63%),电子由HOMO和HOMO-1轨道跃迁到LUMO和LUMO+1轨道.Ni和Fe离子由于有单电子存在,其激发电子跃迁较为复杂.在TPP-Ni中,Ni2+对分子的HOMO轨道有主要贡献,卟啉共轭结构和Ni2+均对LUMO轨道有贡献,TPP-Ni的S0→S1跃迁组成为HOMO-4→LUMO+2(100%),意味着处于较低能量HOMO-4轨道的电子也易被激发跃迁.激发后,电子传递方向是电子从金属向配体的转移,卟啉配体吸收电子后分子结构和光学性质发生变化,能量累积后其稳态吸收光谱增加.TPP-FeCl的HOMO轨道位于Fe和卟啉内部的4个吡咯环上,LUMO则基本位于Fe上,表明Fe在分子中既可以提供空轨道,又可提供电子.TPP-FeCl的电子跃迁更为复杂,其S0→S1跃迁组成主要为HOMO-2(A)→LUMO(A)(19%),HOMO-19(B)→LUMO+2(B)(11%)和HOMO-2(B)→LUMO+2(B)(53%)等;S0→S2跃迁组成主要为HOMO-6(B)→LUMO+2(B)(31%),HOMO-2(B)→LUMO(B)(22%)和HOMO-2(B)→LUMO+3(B)(32%)等,表明铁配位卟啉有更多能级较低的占据轨道电子易被激发,同时Fe也提供更多能级较高的空轨道参与,分子内部电子跃迁途径更为复杂,光激发后,位于铁和吡咯环HOMO轨道上的电子以及更低能级占据轨道电子迅速自由离域至Fe提供的最低空轨道上,电子传递方向是电子从配体向金属的转移,激发态对整体有机分子结构影响很小,因而稳态吸收光谱变化不大,以上分析结果与实验结果相吻合.

Fig.5 Frontier orbital energy levels diagram of TPP-2H
结合稳态吸收光谱、荧光光谱、瞬态吸收光谱、动力学数据及理论计算结果,可以推测出激光激发各卟啉光谱变化的可能机理(图6).TPP-2H的卟啉环中心无金属配体,在稳态下是一个平面结构.TPP-2H受激光激发之后的稳态吸收谱图并未出现新的峰,表明分子结构较稳定;瞬态吸收谱图中出现新峰表明卟啉被激发至激发态.TPP-2H环中心未配位的金属提供空轨道,分子被激发至激发态后电子能量不能快速释放,产生长时中间体,然后再逐渐恢复至稳态.
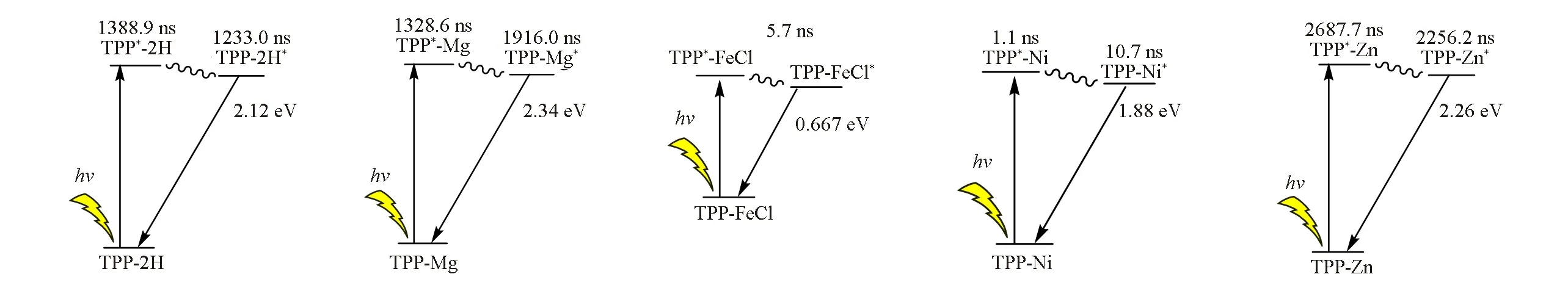
Fig.6 Proposed energy transfer schemes of five porphyrin compounds after the photoexcitation
TPP-FeCl*呈现出的特殊性是因为铁离子的复杂电子排布方式以及与卟啉环复杂的配位,中心三价铁最外层电子轨道为3d5半满结构,基态光谱项为6S,d轨道有5个自旋平行未成对电子,配体不仅具有σ轨道(sp2杂化轨道)而且还有含孤对电子的π轨道(Pz轨道)[25,26].计算结果表明,LUMO轨道位于Fe上,Fe离子由于缺乏电子,配位后可以为分子提供空轨道,易于产生电子π-d跃迁.光激发分子后,TPP*-FeCl中位于卟啉配体的HOMO或更低能级的占据轨道电子迅速自由离域至Fe提供的空轨道上,激发态电子在铁离子中发生辐射或非辐射衰减,使得TPP-FeCl在光激发后相对于另外3种金属卟啉化合物具有良好的受光激发后的稳定性,激发后瞬态恢复到稳态时间极短.激光激发实验表明,在300次连续激光激发下铁卟啉的紫外-可见吸收光谱未发生明显变化,表明铁卟啉在光照条件下稳定性高,不易受光照损坏,对铁卟啉作为肌红蛋白和细胞色素p450等蛋白活性中心的选择有着非常重要的作用.
二价镍离子的最外层电子排布为3d8,与4个氮配位后达到16电子配位稳定结构,镍卟啉的Q带有明显吸收,镍离子基态光谱项为3F,d轨道有2个自旋平行未成对电子.Ni2+对分子的HOMO轨道有主要贡献,卟啉共轭结构和Ni2+均对LUMO轨道有贡献,电子的π-π*跃迁和π-d跃迁均易产生,金属电子也可激发到金属空d轨道上,能量得以从金属离子中发生辐射或非辐射衰减,同时配位结合能力加强.经激光激发,卟啉吸收能量到激发态TPP*-Ni,能量转移后整个激发态分子TPP-Ni*可较迅速衰减至基态.
二价锌离子的最外层电子排布为3d10,电子全满,配位后能形成18电子配位稳定结构,锌离子无未成对电子和d空轨道;镁离子同样外层电子全满,二者均属于闭合壳层,电子占据轨道成为卟啉中心电子传递的缓冲区域.经激光激发,卟啉吸收能量到激发态TPP*-Mg或TPP*-Zn,HOMO和LUMO轨道未分布在金属离子上,电子只能通过π-π*跃迁至卟啉的更高能级轨道,卟啉结构会有所形变,产生长时中间态,能量转移后,整个分子呈激发态的TPP-Mg*或TPP-Zn*再进一步衰减至基态.经过多次激光激发,长时中间态不能及时衰减,有一部分不能恢复至基态,持续吸收光子,金属离子缓冲区域能量过高,超出其能量阈值后则导致分子结构无法保持原构型.以上TPP-Mg或TPP-Zn激发态特性表明,在金属离子缓冲区域存在下两者可产生长时激发中间体,设定特定激发脉冲时间可使得二者在光电转换效率方面发挥独特优势.这一特性可进一步用于阐释自然界光合作用中叶绿素选择镁离子配位的原因,同时本文研究可用以阐明锌卟啉在光电转换方面具有极大优势的原因[27,28].
3 结 论
以四苯基卟啉为实验模板,结合稳态吸收光谱、荧光光谱、瞬态吸收光谱、动力学数据及理论计算结果,发现TPP-FeCl在Soret带和Q带谱峰强度降低均不明显,TPP-FeCl*的瞬态吸收光谱中Soret带的ΔOD值为0,即无明显谱峰,显示Fe与卟啉环的配位较为稳定,且不易受到激光激发影响,相对于其它卟啉化合物具有良好的受光激发后的稳定性,对铁卟啉作为生物蛋白活性中心有着非常重要的作用.TPP-Ni经光激发之后,Soret带和Q带吸收强度均出现大幅上升,显示了其独特光激发性质,原因可能在于Ni能同时提供空d轨道和电子,能量转移后整个激发态分子TPP-Ni*可较迅速衰减至基态.TPP-2H,TPP-Mg和TPP-Zn内部结合的离子外层电子呈饱和状态,产生长时激发态中间体,有利于光电转换或者光化学反应过程,良好的电子转移特性可能是镁卟啉被选择作为叶绿素的活性中心,锌卟啉在太阳能敏化剂等领域发挥着重要作用的原因之一.
猜你喜欢
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
汕头大学学报(自然科学版)(2020年4期)2020-12-14
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
原子与分子物理学报(2015年3期)2015-11-24
读写算·教研版(2014年12期)2014-09-01
